










Molekulárni biológovia pomáhajú v boji s infekčnými chorobami
| 2. 2. 2003Známy je prípad amerického zubného lekára, ktorého podozrievali, že nakazil svojich pacientov nebezpečným HIV-1 spôsobujúcim AIDS; toto podozrenie však bolo treba dokázať, alebo ho naopak vyvrátiť. V súvislosti s výskytom klasického moru ošípaných na Slovensku bolo dôležité zistiť, o aký vírus ide v našich chovoch a v priľahlej geografickej oblasti. Ukazuje sa, že aj k riešeniu takýchto problémov môžu výrazne prispieť molekulárno-genetické metódy.
V molekulárnej epidemiológii a epizootológii sa využívajú poznatky získané klasickým výskumom situácie v teréne a štúdiom genetickej variability kmeňov mikroorganizmu izolovaných z ohnísk nákazy. Využíva sa pritom poznatok, že genóm mikroorganizmu akoby si „pamätal“ históriu svojho vývoja v danom prostredí.
Genetická variabilita mikroorganizmov
Najčastejšie vzniká genetická variabilita mikroorganizmov v dôsledku mutácie nukleotidov (ale aj výmeny časti genómu a iných zmien). Genóm mikroorganizmu nie je konštantný, v prostredí, kde sa množí, prebiehajú mutácie nukleotidov. Mutácie sú v genóme RNA asi o 3–4 rady častejšie ako v genóme DNA. Hoci sa vyskytujú náhodne pozdĺž celého genómu, tieto náhody majú svoje limity. Pretože pri niektorých mutáciách mikroorganizmus hynie, v prírode pozorujeme iba také, ktoré neovplyvnia jeho životaschopnosť. Výsledkom je konštatovanie, že v istom konkrétnom prostredí vznikajú v genóme mikroorganizmu charakteristické mutácie.Analýza genómu mikroorganizmu
Pre presnejšiu typizáciu a určovanie vzťahov medzi ohniskami nákazy je dôležité, aký mikroorganizmus je pôvodcom nákazy. Jeho genóm môžeme analyzovať viacerými metódami, napr. normálnou alebo pulznou gélovou elektroforézou, štiepením genómu reštrikčnými endonukleázami alebo modifikovanou metódou PCR (random PCR). V súčasnosti sa ako najpresnejšia analýza používa sekvenovanie nukleových kyselín, t. j. prečítanie genetickej informácie. S obľubou sa pritom využíva získavanie špecifických fragmentov DNA pomnožením priamo z genómu mikroorganizmu metódou PCR (bližšie pozri Vesmír 74, 134, 1995/3).Základný metodický prístup používaný v molekulárnej epidemiológii a epizootológii
Pretože genetická analýza je náročná na čas i financie, treba k nej pristupovať uvážene. Prvým krokom je dôsledné zhodnotenie epidemiologickej, resp. epizootologickej situácie v teréne. Z doterajšej praxe vyplýva, že pri genetickej analýze mikroorganizmu spravidla stačí sekvenovať úsek 200–500 nukleotidov, čiže netreba analýzovať celý gén alebo genóm. Jednak sa tým skráti čas analýzy, jednak sa znížia náklady. Na porovnanie a potvrdenie výsledkov sa odporúča urobiť genetickú analýzu v dvoch nezávislých oblastiach genómu.Porovnávajú sa nukleotidové sekvencie získané z kmeňov izolovaných z rôznych ohnísk nákazy, pričom sa využívajú rôzne počítačové programy. Výsledkom fylogenetickej analýzy je fylogenetický strom, ktorý grafickou formou vyjadruje vzťahy medzi analyzovanými nukleotidovými sekvenciami. Pretože kmene sú izolované z ohnísk nákazy, fylogenetický strom vyjadruje aj vzťahy medzi ohniskami nákazy. Pri genetickej typizácii mikroorganizmu sa do fylogenetickej analýzy zahŕňajú aj dobre charakterizované referenčné kmene.
Nukleotidové sekvencie, ktoré patria jednotlivým kmeňom, sú na fylogenetickom strome zoskupené do fylogenetických vetiev (línií). Počítačové programy využívajú rôzne algoritmy a modely evolúcie, preto aj fylogenetické stromy môžu mať rôzne tvary a zoskupenie analyzovaných kmeňov v nich môže byť podobné alebo aj odlišné. Fylogenetický strom skonštruovaný počítačovým programom je len matematickou aproximáciou vzťahov medzi analyzovanými nukleotidovými sekvenciami, preto treba venovať veľkú pozornosť a starostlivosť jeho interpretácii.
V konečnej fáze analýzy sa opäť spája úsilie epidemiológa alebo epizootológa a molekulárneho biológa. Pri interpretácii fylogenetického stromu sa berie do úvahy celková nákazová situácia v teréne. Len symbiózou poznatkov z dvoch rôznych, doteraz nezávislých odborov možno dosiahnuť výsledky využiteľné v praxi. Úloha epidemiológa či epizootológa je však v tomto procese nezastupiteľná, aby sa analýza nestala len akousi počítačovou hrou neodrážajúcou reálne problémy v teréne.
Riešenia niektorých praktických problémov vírusových nákaz
V prvej polovici 90. rokov sa vyskytlo v oblasti Four Corners (Nové Mexiko, Colorado, Arizona, Utah) v USA nebezpečné ochorenie dýchacích ciest (pulmonárny syndróm) s vysokou úmrtnosťou ľudí stredného veku. Prvé analýzy naznačili, že infekčným činiteľom by mohol byť hantavírus prenášaný hlodavcami. Na veľké prekvapenie vedcov vírusové sekvencie získané z ľudských pacientov sa nezaradili k známym hantavírusom, ale vytvorili na fylogenetickom strome novú vetvu, ktorá usvedčila vírus ako nový hantavírus, neskôr pomenovaný ako Sin Nombre vírus.Nukleotidové sekvencie vírusov z hlodavcov a ľudských pacientov z rovnakého ohniska nákazy boli veľmi podobné, čo svedčilo o prenose vírusu z hlodavcov na človeka. Naproti tomu vírusové sekvencie získané z ohnísk v Arizone, Colorade a Novom Mexiku boli rozdielne. U jedného pacienta z Arizony však zistili vírus, ktorý sa nepodobal ostatným miestnym vírusovým kmeňom, ako sa všeobecne očakávalo, ale skôr vírusom zisteným v Colorade. Z podrobnejšieho epidemiologického zisťovania vyplynulo, že pacient bol infikovaný počas krátkeho pobytu v Colorade. Genóm tohto vírusu vykazoval charakteristické znaky „coloradského typu“, teda „pamätal si“ históriu svojho vývoja. 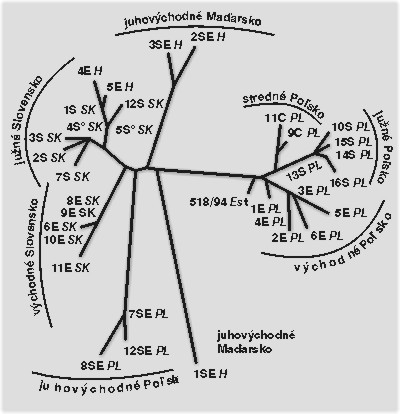
V prípade v úvode spomínaného amerického lekára obvineného z nakazenia svojich pacientov vírusom HIV-1 bolo treba nájsť exaktný dôkaz. Vírus izolovaný z lekára, inkriminovaných pacientov a na porovnanie aj z niektorých miestnych obyvateľov infikovaných HIV-1 čiastočne sekvenovali a skonštruovali fylogenetický strom. Z genetickej analýzy vyplynulo, že vírusy infikovaných pacientov a lekárov vírus boli takmer zhodné, ale odlišné od ostatných vírusových kmeňov. To napokon usvedčilo lekára.
Spomínané metódy sa využívajú aj v molekulárnej epizootológii pri analýze šírenia nebezpečných nákaz zvierat. Medzi takéto nákazy patrí tiež klasický mor ošípaných, ktorý sa monitoruje na medzinárodnej úrovni. Ohniská tohto ochorenia na Slovensku sa začali objavovať vo väčšom počte od konca roku 1993, keď sme sa prispôsobili krajinám EÚ a upustili sme od očkovania. Pripomeňme, že v prípade výskytu nákazy sa spravidla všetky zvieratá z inkriminovaného chovu utratia – na zamedzenie jej šírenia. Napr. koncom 90. rokov v Holandsku takto utratili takmer desať miliónov ošípaných. V súvislosti s ohniskami nákazy na Slovensku nás zaujímalo, aký vírus sa vyskytuje v našich chovoch a v priľahlej geografickej oblasti.
Vírusové kmene izolované na Slovensku patrili do skupiny európskych kmeňov. Zoskupovanie kmeňov vírusu klasického moru ošípaných na fylogenetickom strome prísne sledovalo ich geografický pôvod (viď obrázek). Charakteristické skupiny vytvorili vírusové kmene izolované z ohnísk na južnom a východnom Slovensku. Na fylogenetickom strome sa našli medzi kmeňmi z južného Slovenska aj niektoré kmene z Maďarska. Poukazovalo to na možné zavlečenie nákazy, ale fylogenetický strom neukáže smer prenosu. Možno to určiť opäť iba podrobnejším epizootologickým prieskumom v teréne. Osobitnú fylogenetickú líniu tvorili aj kmene z juhovýchodného Poľska, ďalšie dve predstavovali kmene zo stredného a východného Poľska, oddelené línie tvorili aj kmene z Maďarska. Porovnaním českých a slovenských kmeňov sme zistili, že sú rovnaké alebo veľmi podobné, čo opäť poukazovalo na ich spoločný pôvod.
Získaná databáza nukleotidových sekvencií kmeňov vírusu klasického moru ošípaných (ale principiálne aj z kmeňov akéhokoľvek mikroorganizmu) spolu s databázou sekvencií z kmeňov získaných z iných častí Európy a sveta je neobyčajne užitočná pri analýze budúcich ohnísk tejto nákazy. V prípade zaregistrovania nového ohniska stačí rýchlo sekvenovať časť vírusového genómu z jedného-dvoch kmeňov, sekvenciu vložiť do databázy analyzovaných kmeňov a urobiť fylogenetickú analýzu. Z polohy vírusového kmeňa na fylogenetickom strome možno ľahšie určiť pôvod nákazy.
Veľmi pozorne sa sleduje opätovné zavlečenie veľmi nebezpečnej slintačky a krívačky (food-and-month disease – FMD) na európsky kontinent, zvlášť na Britské ostrovy (začiatkom roku 2001). Epizootológov, prirodzene, zaujíma pôvod tejto nákazy zvierat. Za týmto účelom boli čiastočne sekvenované vírusové kmene izolované v ohniskách nákazy v Anglicku. Z fylogenetickej analýzy vyplynulo, že vírus FMD zachytený v Anglicku nie je originálny, ale veľmi podobný vírusom detegovaným predovšetkým v ázijských štátoch. Tento nebezpečne sa šíriaci vírusový kmeň patrí k serotypu „O“ a vzhľadom na jeho široký geografický výskyt bol nazvaný panázijský kmeň. Výsledok analýzy naznačuje, že vírus bol do Veľkej Británie zavlečený z Ázie. Presný spôsob zavlečenia nákazy je predmetom výskumu klasickej epizootológie.
Opísaný molekulárno-genetický prístup má širšie využitie. Možno ho využiť v epidemiológii a epizootológii na určovanie vzťahov medzi ohniskami nákazy, na presnú typizáciu kmeňov rôznych mikroorganizmov, pri diferenciácii vakcinačných kmeňov, pri potvrdzovaní objavov nových mikroorganizmov. Tento postup sa využíva aj pri cielenejších taxonomických analýzach a štúdiu evolúcie mikroorganizmov.
Ke stažení
 Článek ve formátu PDF [594,41 kB]
Článek ve formátu PDF [594,41 kB]
O autorovi
Štefan Vilček

Doporučujeme

Když bahno teče jako ledovec

Ideologie v mapách, mapy v rukách ideologů 
