










Bioinformatika v léčbě rakoviny
Tradiční postupy léčby onkologických onemocnění se sice stále zdokonalují, ale často nejsou selektivní – postihují i zdravé buňky a tkáně, což vede k řadě nepříjemných, až život ohrožujících vedlejších účinků. I přes značné pokroky navíc nejsou tyto metody účinné vždy. U některých pacientů může rakovina recidivovat nebo se stát vůči léčbě rezistentní. Naštěstí se v posledních letech objevuje nová naděje v podobě personalizované medicíny, jejíž součástí je i precizní medicína. Jak jí pomáhá bioinformatika?
Cílem precizní medicíny je vybrat tu nejvhodnější léčbu pro každého pacienta s ohledem na jeho specifické potřeby a vlastnosti nádoru. To umožňuje efektivnější a šetrnější léčbu s menším počtem vedlejších účinků a s větším potenciálem pro trvalé vyléčení.
Zejména rozvoj vysokopropustného sekvenování (next-generation sequencing, NGS) umožňuje rychlé a nákladově efektivní molekulární profilování nádoru, a tedy identifikaci specifických mutací a biomarkerů, které ovlivňují jeho chování a citlivost na léčbu. Tyto informace pak může využít molekulární tumorová komise (viz s. 636), která navrhne optimální léčbu. Rozvoj NGS v klinické onkologii nicméně přináší také výzvy ve formě obrovského množství dat, která je kvůli inherentnímu šumu a přirozené genetické variabilitě pacientů složité analyzovat. Klíčovou roli proto hraje bioinformatika.
Role v precizní onkologii
Bioinformatika umožňuje vědcům a lékařům analyzovat a interpretovat obrovské množství genomických dat o nádorech a pacientech získaných z NGS. Pomocí sofistikovaných výpočetních nástrojů a algoritmů pomáhá odhalit relevantní informace o mutacích, molekulárních profilech a imunitním stavu nádoru, a poskytuje tak lékařům cenné informace pro rozhodování o léčbě.
Konkrétně se bioinformatika uplatňuje v následujících oblastech:
1) Analýza sekvenačních dat včetně sestavení genomu nebo transkriptomu, anotace genů a regulačních oblastí nebo analýza exprese genů.
2) Srovnávací analýza genů, identifikace mutací a konzervovaných sekvenčních oblastí, které jsou často klíčové pro funkci genů nebo regulaci jejich exprese.
3) Strukturní analýza proteinů a vliv mutací na jejich stabilitu, funkci a schopnost interagovat s dalšími proteiny v rámci regulačních a metabolických drah.
4) Hledaní nových léčiv a nového využití těch známých pomocí virtuálního screeningu obrovských databází malých chemických sloučenin.
5) Vizualizace dat pro snadnou a srozumitelnou interpretaci.
6) Vývoj databází pro uchovávání dat, jejich sdílení a snadný a přehledný přístup k již známým informacím.
V posledních letech se v onkologii čím dál více využívají také přístupy strojového učení a umělé inteligence (AI) – jak samostatně, tak v kombinaci právě s bioinformatikou. Umožňuje to lepší diagnostiku při analýze snímků ze zobrazovacích technik (rentgenového vyšetření, magnetické rezonance, výpočetní tomografie nebo mamografie). Tyto přístupy umožňují identifikaci nádorů s větší přesností a efektivitou než lidské oko (Vesmír 103, 392, 2024/7). AI se začíná využívat i při vývoji diagnostických testů, analýze genomických dat, sledování pacientů, hledání nových léčiv, prognóze reakcí na chemoterapii. Vzhledem k rychlému vývoji AI je pravděpodobné, že se její využití v onkologii v příštích letech ještě rozšíří.
Software PredictONCO
Díky pokročilým výpočetním metodám je možné ze sekvencí mutovaných proteinů získávat mnohem více informací o vlivu na strukturu proteinu, jeho stabilitu, funkci a interakce s dalšími proteiny i malými molekulami. Výpočetně predikované a pečlivě interpretované výsledky mohou napomoci k rychlým a dobře informovaným rozhodnutím. Pokud se výpočty provádějí za stejných podmínek na původním i mutovaném proteinu, je možné provést komparativní analýzu. Provedení všech zmíněných analýz nicméně vyžaduje značné znalosti výpočetní biologie a bioinformatiky. Navíc by manuální provedení analýz bylo velmi zdlouhavé a neumožňovalo by analýzu mutací u většího množství pacientů.
Proto byl ve skupinách Davida Bednáře a Stanislava Mazurenka v Loschmidtových laboratořích na Přírodovědecké fakultě Masarykovy univerzity vyvinut PredictONCO, webový server pro plně automatizovanou a rychlou analýzu vlivu mutací na funkci proteinů relevantních pro onkologii.1) PredictONCO využívá metody molekulového modelování, bioinformatické analýzy a strojového učení. Během několika dnů generuje podrobné zprávy o efektu analyzovaných mutací na funkci proteinů.
Práce v PredictONCO probíhá ve dvou hlavních krocích: 1. výběr cílového proteinu a mutace a 2. analýza dat.
Výběr proteinu a mutace
V prvním kroku si uživatel vybírá, který z 44 předem definovaných proteinů souvisejících s onkologií ho zajímá. Tyto proteiny byly vybrány odborníky s cílem pokrýt většinu běžných onkogenních spouštěčů a terapeutických cílů. Systém zatím nepodporuje nahrávání vlastních proteinových struktur. Důvodem je riziko chybné biologické interpretace získaných výsledků. Většinu proteinů relevantních pro onkologii totiž představují komplexní transmembránové proteiny, pro které není k dispozici jediná spolehlivá experimentální struktura vhodná pro analýzy bez rozsáhlého odborného zpracování. Pokud je však zapotřebí přidat nový protein do seznamu cílů, uživatelé tak mohou učinit přes webové rozhraní PredictONCO. Po výběru proteinu je zadána mutace buď napsáním substituce, nebo výběrem pozice nahrazované aminokyseliny z vizualizované sekvence proteinu.
Výpočty obvykle trvají do 48 hodin, v závislosti na délce proteinu a dalších strukturních parametrech. Tento časový limit byl zvolen jako kompromis mezi rychlým poskytnutím dat pro včasnou léčbu a snahou využít co nejspolehlivější a nejpřesnější výpočetní metody.
Analýza dat
Již hotové výpočty jsou uloženy v databázi a při požadavku na dříve spočítanou mutaci je výsledek okamžitě načten z uložených dat. V případě nové mutace je provedeno několik analýz. Analýza struktury (pokud je k dispozici vhodná 3D struktura) se využije k posouzení možných disruptivních změn ve struktuře a k provedení komparativní analýzy oproti původní struktuře. Je predikován vliv mutace na stabilitu proteinu, funkci a vazbu léčiv (pouze pro mutace v katalytickych doménách). Následně je proveden virtuální screening rozsáhlé databáze léčiv schválených americkým regulátorem FDA a evropskou lékovou agenturou EMA pro nalezení potenciálních inhibitorů. Analýza sekvence pak umožňuje predikci vlivu mutace na funkci proteinu a analýzu konzervovanosti na základě příbuzných proteinů. Dále jsou extrahovány relevantní informace z veřejně dostupných databází.
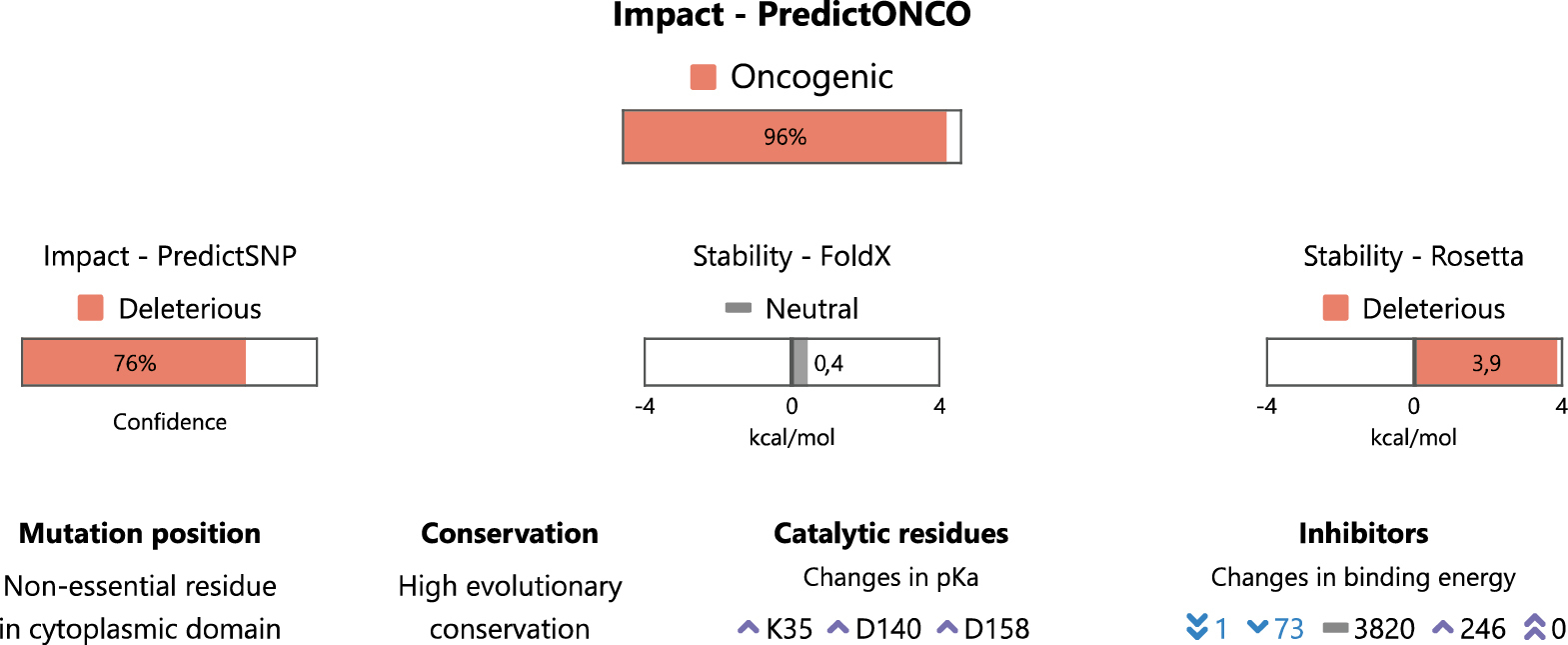
Všechny získané hodnoty z analýz struktury a sekvence se následně využijí jako vstupy pro nově vyvinutý prediktor založený na strojovém učení. Ten rozhodne, zda mutace vede k rozvoji onkologického onemocnění, a tudíž je potřeba se tímto poškozeným proteinem dále zabývat, nebo zda jde o mutaci neškodnou, a tedy pro následnou léčbu nepodstatnou. Jistota predikce je vyjádřena konfidenčním intervalem (0–100 %). Všechny důležité informace jsou pak prezentovány v sekci Summary (obr. 1).
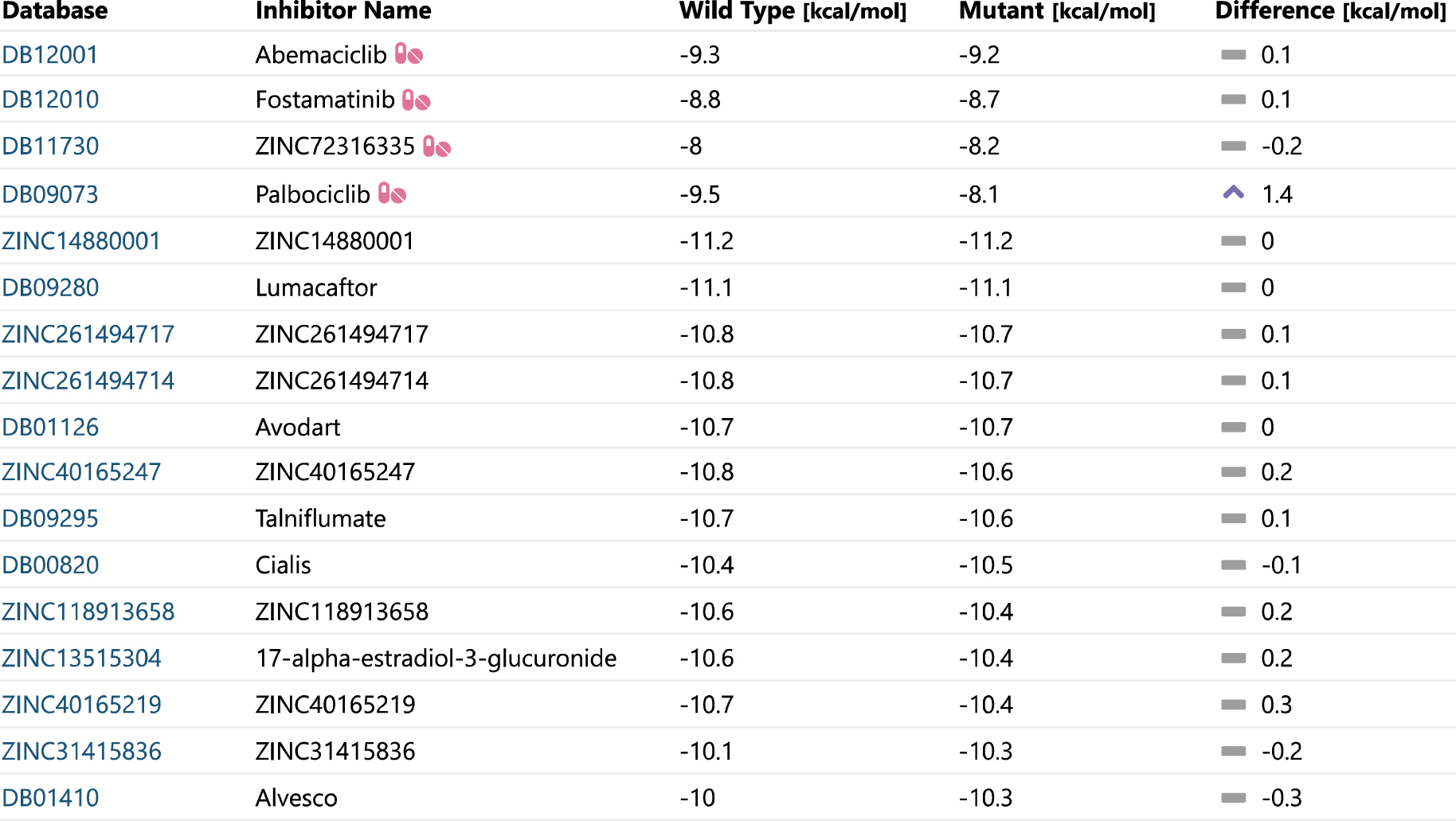
Pod sekcí shrnující nejdůležitější výsledky se nacházejí podrobné informace z jednotlivých predikcí, které uživatel může využít pro detailní analýzu dané mutace. Konkrétně to jsou informace o mutantní aminokyselině, katalytických aminokyselinách, analýze konzervovanosti, 3D vizualizaci proteinu, screeningu inhibitorů a další informace z databází. Velmi důležitá je sekce Inhibitors (obr. 2), která zobrazuje vazebné energie všech potenciálních léčiv schválených organizacemi FDA a EMA. Tabulka je řazena tak, aby nejdříve poskytovala informace o lécích, které jsou využívány v souvislosti s cílovým proteinem (označeny speciální ikonou). Následují ostatní látky, které vykazují nejlepší vazbu a mají potenciál být účinným léčivem.
Úspěšnost predikce a další vývoj
Nástroj PredictONCO byl trénován na více než tisícovce známých mutací. Testování proběhlo na nezávislé sadě 216 mutací pro sekvenční a 89 mutací pro strukturní prediktor. Výsledná přesnost byla vynikající, PredictONCO překonal všechny testované metody včetně nejnovějšího nástroje ESM Variants.
Současná verze však má i několik omezení. Server sice dokáže identifikovat mutace způsobující ztrátu funkce, ale nemůže na jejich základě navrhnout léčebný postup. Proto je v plánu začlenit znalosti o biologických drahách, což umožní inhibici proteinů nacházejících se níže v dané dráze. PredictONCO také neumí předpovídat účinky mutací způsobených inzercemi a delecemi v proteinové sekvenci, ačkoliv i tyto mutace jsou často důležité pro rozvoj onemocnění. Tento problém zatím bohužel není vyřešen obecně a neexistují žádné spolehlivé nástroje. Věříme však, že s vývojem nových metod se i tyto mutace podaří do analýzy zahrnout.
Další nevýhodou je omezený počet analyzovaných proteinů. Aktuálně se snažíme jejich seznam rozšířit o jiné klinicky zajímavé cíle, nicméně s nedostatkem kvalitních struktur je současný postup pouze poloautomatický, a proto pomalý. Pokroky v oblasti strukturní biologie by však mohly tento problém do budoucna vyřešit. Zejména vývoj nástrojů AI jako AlphaFold (Vesmír 103, 326, 2024/6) má velký potenciál v nejrůznějších odvětvích včetně precizní onkologie.
Poznámky
1) PredictONCO je volně přístupný na https://loschmidt.chemi.muni.cz/predictonco, kde je k dispozici i podrobný manuál. Popis nástroje i příkladové analýzy jsou zveřejněny v publikaci Stourac J. et al.: Briefings in Bioinformatics 25, 1, bbad441, 2023, DOI: 10.1093/bib/bbad441.
Ke stažení
 článek ve formátu pdf [620,72 kB]
článek ve formátu pdf [620,72 kB]
O autorovi
David Bednář

Doporučujeme

Když bahno teče jako ledovec

Ideologie v mapách, mapy v rukách ideologů 
