









Neurodegenerace
Stárnutí populace v rozvinutých státech Evropy i Ameriky, hrozba „nemoci šílených krav“ či nové formy Creutzfeldtovy-Jakobovy choroby a náhlý rozvoj neurobiologie – to vše zvýšilo zájem o příčiny neurodegenerativních chorob (viz rámeček). Jejich společným rysem je zánik některých populací nervových buněk, který se projevuje jak neurologickými, tak psychiatrickými příznaky. K těm prvním patří poruchy koordinace pohybu, změny svalového napětí, poruchy hybnosti a mimovolní pohyby, k druhým poruchy paměti, poruchy kognitivních funkcí přecházející postupně v demenci, kvalitativní poruchy vědomí (např. delirium), poruchy nálady či změny osobnosti, které bývají zcela nepochopitelné pro pacientovo okolí. Závažnost klinických příznaků jednotlivých onemocnění zpravidla (ne vždy) odpovídá množství „ztracených“ nervových buněk či míře jejich postižení. Ačkoliv se jednotlivé neurodegenerativní choroby od sebe značně liší, všechny se vyznačují progresivním rozvojem a téměř úplnou bezmocností současné medicíny v jejich léčbě. Teprve v posledních několika letech se dovídáme bližší informace o molekulární podstatě těchto chorob (viz též J. Horáček, L. Motlová, Vesmír 78, 307, 1999/6). Jak se v poslední době ukazuje, velká část těchto nemocí je způsobena „špatně sbalenými“ proteiny, ukládanými postupně v buňkách centrální nervové soustavy. Tato porucha spojuje i tak různorodé choroby, jako jsou Huntingtonova chorea, Parkinsonova choroba nebo Alzheimerova nemoc.
Jak „špatné sbalení“ vzniká? Proteiny se skládají z řetězců aminokyselin. Můžeme si je představit jako korálky na šňůře, která je složitým, ale poměrně přesně definovaným způsobem smotaná do určitého obrazce. Jestliže se „pomuchlá“ (složí nesprávně), náhrdelník se nedá nosit (protein není funkční). „Korálků“ (aminokyselin) máme přesně dvacet. Pořadí, v němž jsou na „šňůře“ (polypeptidovém řetězci) poskládány, spolurozhoduje o výsledné prostorové struktuře proteinu, a tím i o jeho funkci. O tom, jestli se protein sbalí do správné, biologicky aktivní konformace, rozhodují vedle pořadí aminokyselin další vlivy, jako je přítomnost pomocných bílkovin, chaperonů („gardedám“ nově vytvářených proteinů), stopových prvků, sacharidových molekul na povrchu nově vytvořeného proteinu ad.
Geneticky podmíněná neurodegenerace
Pořadí aminokyselin v peptidovém řetězci je určeno pořadím párů bází v molekule DNA. Někdy však vznikne mutace DNA s takovým pořadím aminokyselin, že se nová bílkovina špatně rozpouští, popřípadě začíná polymerovat. Přesně to se děje u Huntingtonovy chorey a řady dalších neuropatií: v genu lokalizovaném na 4. chromozomu a kódujícím protein huntingtin se vyskytuje nestabilní sekvence opakující triplet CAG, což je nukleotidový kód pro aminokyselinu glutamin. Jestliže je takových tripletů zřetězeno více než obvyklých 6–37, vzniká bizarní, biologicky zřejmě nefunkční, špatně rozpustná bílkovina (viz též Vesmír 77, 15, 1998/1), což má za následek vývoj nerozpustných plaků, neurodegeneraci a buněčnou smrt.Obdobný mechanizmus (pomalé ukládání špatně sbalených proteinů) se skrývá i za dalšími geneticky podmíněnými neurodegenerativními chorobami, jako jsou Parkinsonova choroba, amyotrofická laterální skleróza nebo Alzheimerova choroba. Chybně sbalenými proteiny, jejichž ukládání vede k neurodegeneraci, může být cytoskeletární protein tau, enzym superoxiddismutáza, membránově vázaný amyloidový prekurzorový protein ad.
Priony
Pokud je choroba podmíněna geneticky, tj. chybné složení proteinu je dáno nesprávnou sekvencí aminokyselin (například dlouhými řetězci polyglutaminů), zdá se všechno jasné. Co si však počít s případy, kdy není žádné genetické podmínění zjištěno, příslušné geny mají „normální“ sekvenci, je však podezření na infekční přenos, jako je tomu u choroby kuru, bovinní spongiformní encefalopatie nebo nové formy Creutzfeldtovy-Jakobovy nemoci? Vypadá to, že se některé proteiny mohou sbalovat do dvou forem, z nichž jedna odpovídá normální funkční bílkovině, druhá je nerozpustná a snadno tvoří agregáty. Chybně sbalený protein může vyvolat změnu struktury správně sbalených proteinů (jak to dělá, to nevíme) , což postupně vede k tvorbě nerozpustných plaků a k neurodegeneraci. V tomto smyslu jsou špatně sbalené proteiny „infekční“ – odtud jejich jméno „priony“ (více o prionech v článku P. Buška, Vesmír 81, 14, 2002/1).
, což postupně vede k tvorbě nerozpustných plaků a k neurodegeneraci. V tomto smyslu jsou špatně sbalené proteiny „infekční“ – odtud jejich jméno „priony“ (více o prionech v článku P. Buška, Vesmír 81, 14, 2002/1).
Co si však počneme s neurodegenerativní chorobou, která není nutně podmíněna geneticky, a přitom u ní nebyla prokázána role prionů?
Alzheimerova choroba
V mozcích pacientů trpících Alzheimerovou chorobou se vyskytují dva různé proteinové agregáty: smotky chybně složeného cytoskeletárního proteinu tau a plaky z proteinových agregátů tvořených fragmenty membránového proteinu: β-amyloidového prekurzoru (β-APP). Přítomnost dvou druhů proteinových agregátů vedla ve vědecké obci k prudkým sporům o to, který z těchto proteinů je skutečnou příčinou onemocnění a který jen vedlejším příznakem. Zastánci jednotlivých teorií (posměšně nazývaní tauisté a baptisté) jdou ve svém nadšení pro oblíbenou teorii někdy tak daleko, že se v přednáškách na velkých vědeckých konferencích o protivném proteinu vůbec nezmiňují. Podle posledních prací (Science 293, 1487, 2001) se ale zdá, že oba mechanizmy jsou vzájemně provázány. Vzhledem k tomu, že autorem tohoto článku je přesvědčený baptista, neuslyšíte o proteinu tau v dalším textu už ani slovo.β-APP je transmembránový protein nejasné biologické funkce (viz obr.1). Blízko N-konce tohoto proteinu se vyskytuje sekvence 42 aminokyselin zvaná amyloidový β-peptid. Právě tento fragment totiž vytváří ony proteinové agregáty nalézané v mozcích pacientů při Alzheimerově nemoci. Tento peptid snadno vytváří fibrilární agregované struktury, které jsou neurotoxické a vedou k vývoji této choroby.
„Hodné“ a „zlé“ proteázy
Jako téměř každý protein je i β-APP degradován proteázami. Jde o enzymy schopné více či méně specificky štěpit peptidové vazby, tj. „roztrhnout korálový náhrdelník na menší kusy“. Některé proteázy si místo štěpení nevybírají, jsou schopny hydrolyzovat téměř každou peptidovou vazbu (např. trávicí enzymy pepsin nebo chymozin). Jiné, zajímavější proteázy jsou vybíravější, štěpí velmi specifické sekvence aminokyselin a mohou tak mít regulační roli (např. proteázy retrovirů, viz Vesmír 80, 332, 2001/6)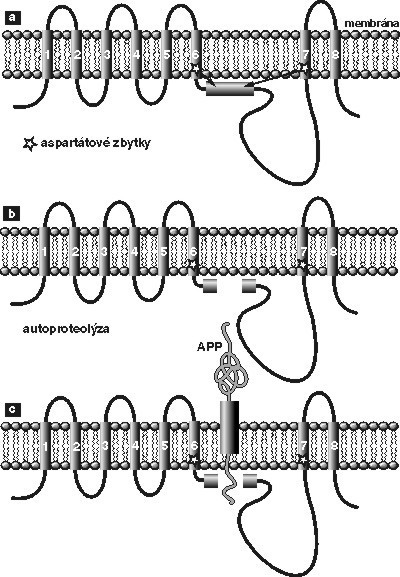 . β-APP je štěpen třemi takovými specifickými proteázami.
. β-APP je štěpen třemi takovými specifickými proteázami.
První z nich, α-sekretáza, štěpí prekurzorový protein právě uvnitř β-amyloidového peptidu za vzniku jednoho rozpustného fragmentu a delšího peptidu, který zůstává zanořen do membrány. Takto rozštěpený protein je „neškodný“, není schopen vytvářet proteinové agregáty; α-sekretáza je tedy „hodná“ proteáza. Další dvě proteázy, štěpící prekurzor nad amyloidovým peptidem a pod ním (viz obr.1), ponechávají amyloidový peptid netknutý. Ten se pak uvolní z prekurzoru a může začít pomalu vytvářet toxické fibrily; β- a γ-sekretázy jsou tedy „zlé“ proteázy. Jejich aktivita vypouští džina amyloidového peptidu z láhve a spouští tak patologický proces, který končí úplným rozpadem osobnosti nemocných Alzheimerovou chorobou.
Identifikace sekretáz
Ještě v r. 1999 končily práce o β-APP smutným konstatováním, že jednotlivé sekretázy dosud nebyly identifikovány. Radikálně se to změnilo během roku 2000. Nejprve byla na dvou amerických pracovištích v tutéž dobu rozpoznána β-sekretáza. Později se ukázalo, že se v lidském genomu vyskytují dvě homologní verze tohoto enzymu, a to BACE1 a BACE2 (z anglického „β-amyloid precursor converting enzyme“). Jde o membránově vázanou aspartátovou proteázu, která svou sekvencí i prostorovou strukturou trochu připomíná trávicí enzymy, jako jsou pepsin nebo chymozin. Ačkoli je tato proteáza schopna uvolnit amyloidový peptid, nezdá se, že má při vzniku Alzheimerovy choroby důležitou roli. Genové mutace nalézané při rané familiární formě této choroby se totiž v oblastech kódujících BACE nevyskytují.- α-sekretáza jakožto „hodný“ enzym budila vždy mnohem menší zájem než „zlé“ proteázy, jak už to v životě chodí. V průběhu roku 2000 se objevilo několik prací, které tento hypotetický enzym identifikovaly jako některou ze známých, membránově vázaných metaloproteáz ADAM 10 a ADAM 17 (metaloproteázy jsou enzymy štěpící peptidovou vazbu, které mají ve svém aktivním místě molekulu kovu, často zinku nebo manganu).
- γ-sekretáza je z těchto nově identifikovaných enzymů nejzajímavější (viz obr.2). Jak je ukázáno na obrázku, tento enzym štěpí β-APP v místě, které je ukryto uvnitř membrány. Donedávna jsme si neuměli představit proteolytický enzym, který by mohl štěpit (hydrolyzovat) peptidovou vazbu v bezvodém prostředí lipidové membrány. Trvalo přes deset let, než se trpělivým studiem mutací vedoucích k rané dědičné formě Alzheimerovy choroby identifikoval protein, o němž se dalo předpokládat, že souvisí s tvořením plaků. Dostal název presenilin. Jak je vidět na obrázku 2, jde o transmembránový protein, který membránu prochází osmi smyčkami. Podle aminokyselinové sekvence nemá tento protein nic společného s žádnou známou proteázou, a proto se donedávna předpokládalo, že se štěpení β-APP účastní jen nepřímo (např. že aktivuje skutečnou γsekretázu). V průběhu posledních dvou let se však objevila série nepřímých důkazů, že dlouho hledanou γ-sekretázou je skutečně presenilin. Ve dvou protilehlých transmembránových smyčkách presenilinu se nacházejí dva aspartátové zbytky (viz obr.2), které svou topologií trochu připomínají aktivní místo aspartátových proteáz, jako jsou pepsin nebo chymozin. Přestože tím analogie končí (na úrovni sekvence aminokyselin žádná podobnost neexistuje a žádná známá aspartátová proteáza není schopna štěpení uvnitř membrány), mutace obou těchto aspartátů opravdu zastavuje γ-sekretázovou aktivitu. To vedlo týmy Christiana Haasse z Mnichova, Denise Selkoe z Harvardu a Sukarto Sinhy ze San Franciska k formulaci představy, že γ-sekretáza je novou aspartátovou proteázou schopnou štěpit uvnitř membrány. Brzo nato se podařilo γsekretázu inhibovat specifickými inhibitory aspartátových proteáz a chemickými metodami se zjistilo, že se tyto inhibitory skutečně vážou mezi dva aspartáty presenilinu. Pokud je tomu skutečně tak a presenilin je opravdu dlouho hledanou γ-sekretázou, řada genetických studií začíná dávat biochemický smysl. Takřka všechny mutace způsobující časnou familiární formu Alzheimerovy nemoci jsou totiž lokalizovány buď poblíž štěpeného místa amyloidového prekurzoru, nebo v genu pro presenilin. Biochemicky řečeno, mutuje buď enzym, nebo jeho substrát. Přestože přímý důkaz podán nebyl a některé výsledky jsou kontroverzní, zdá se, že další otázka byla vyřešena.
Co je to demence?
Demence jsou onemocnění, která se projevují postupnou ztrátou paměti a zhoršením jak poznávacích funkcí (racionálního úsudku, motivací, intelektu), tak dalších (změnou osobnosti, paranoidním myšlením aj.) Nejde tedy o věčné hledání založených brýlí nebo o neschopnost vzpomenout si, jak se jmenuje učitel tělocviku vaší dcery. Úpadek je tak velký, že postižený jedinec není schopen vykonávat běžné denní aktivity, potřebuje asistenci jiné osoby a postupně se stává zcela závislým na svém okolí. Ztráta paměti vede k tomu, že nemocný přestane poznávat své okolí, nepozná ani své nejbližší, a nakonec už neví, kým je on sám. Důsledkem většiny demencí je smrt, a to buď nepřímým působením (např. po infekci či úrazu), nebo přímým vlivem (selháním životních funkcí např. u Alzheimerovy nemoci).
Demence nejsou jen problémem medicínským, ale rovněž (vzrůstajícím) problémem společensko-sociálním. Nezapomeňme, že kvalita péče o dementní a všechny další chronické pacienty je odrazem kulturní vyspělosti národa. Jak jsme na tom my?
NEURODEGENERATIVNÍ CHOROBY ZPŮSOBENÉ UKLÁDÁNÍM NESPRÁVNĚ „SLOŽENÝCH“ PROTEINŮ
(viz rovněž Vesmír 78, 328, 1999/6)
- Alzheimerova choroba je nejčastější ze všech demencí (tvoří zhruba 50–60 % všech případů). Podrobněji se jí zabývá rámeček T. Hájka na protější straně.
- Pickova choroba je velmi vzácná demence, rovněž podobná Alzheimerově nemoci. Atrofie mozkové kůry postihuje hlavně čelní a spánkový lalok, což způsobuje velmi nápadný klinický obraz. V počátečním stadiu choroby se objevují především hrubé změny osobnosti a postižení jedinci se často dopouštějí drobných sexuálních deliktů. Případné trestní stíhání je samozřejmě zastaveno pro nepříčetnost. Atrofie kortexu pokračuje poměrně rychle a nemoc končí smrtí.
- Parkinsonova choroba byla poprvé popsána r. 1817. Je častější u mužů a v pozdějším věku, ale nevyhýbá se ani mladým lidem. Klinicky se projevuje svalovým třesem (tremorem), který je nejvíce patrný v klidu a při pohybu mizí, dále ztuhlostí, pomalostí pohybů, šoupavou chůzí. Deprese se vyskytuje u 30 % pacientů a u 10–30 % se rozvine demence podobná Alzheimerově nemoci. Nemoc je způsobena úbytkem nervových buněk produkujících neurotransmiter dopamin v substantia nigra. Příznaky se objevují při destrukci více než 70 % dopaminergních neuronů v této oblasti mozku.
- Huntingtonova chorea je autozomálně dominantní dědičné onemocnění. Defektní gen leží na 4. chromozomu. Postihuje osoby ve věku 20 až 50 let. Pacienti přežívají průměrně 15 let. Klinicky se projevuje mimovolními pohyby připomínajícími tanec (odtud chorea), psychotickými příznaky (halucinacemi, bludy) a postupující demencí. Takové klinické příznaky jsou důsledkem destrukce malých neuronů ve dvou částech mozku – v ocasatém jádře (nucleus caudatus) a skořápce (putamen).
- Amyotrofická laterální skleróza je v anglosaské literatuře rovněž známa pod názvem Lou Gehrigova choroba (podle slavného hráče na první metě baseballového klubu Yankees, který tímto onemocněním trpěl). Postihuje častěji muže. Klinické příznaky této choroby jsou způsobeny destrukcí velkých motoneuronů, především v předních rozích míšních. To vede k degeneraci laterálních svazků míšních – skleróze – a k následné svalové atrofii. Prvními klinickými příznaky bývá svalová slabost končetin. Atrofie se postupně rozšiřuje i na svaly životně důležité (dýchací a polykací svalstvo) a končí smrtí.
- Creutzfeldtova-Jakobova choroba je infekční prionové onemocnění, ale též geneticky podmíněné. Spongiformní encefalopatie (houbovitý vzhled mozkové tkáně) vede k rychle postupující demenci. Průměrné přežití je šest měsíců.
- Kuru je vzácná infekční prionová choroba vyskytující se na Papui-Nové Guinei u kmene Fore, který holdoval rituálnímu kanibalizmu. Priony se přenášejí především pojídáním mozků a jater zemřelých příslušníků kmene. Klinický i patologický obraz kuru je shodný s Creutzfeldtovou-Jakobovou chorobou.
- Scrapie a bovinní spongiformní encefalopatie (známá veřejnosti jako BSE a ještě častěji jako „nemoc šílených krav“) jsou infekční prionové veterinární formy Creutzfeldtovy-Jakobovy choroby. Scrapie postihuje ovce a kozy, BSE hovězí dobytek. Jde rovněž o chorobu s rychlým postupem zmatenosti a demence.
Alzheimerova demence v kostce
- Epidemiologie: Alzheimerova nemoc je nejčastější degenerativní onemocnění mozku, představuje asi dvě třetiny všech demencí ve stáří (viz též Vesmír 78, 307, 1999/6 a Vesmír 78, 328, 1999/6). Častěji se vyskytuje u žen. Postihuje 5–10 % osob ve věku 65 let a v 80–89 letech jí trpí každý desátý člověk (jen v USA jsou jí postiženy asi čtyři miliony osob).
- Věk: Propuká většinou po 60. roce života a její prevalence (počet nemocných na 1000 obyvatel) s každou další věkovou dekádou exponenciálně narůstá. Jsou však zaznamenány i příznaky nemoci u třicetiletých. Odlišujeme dvě varianty – s časným nástupem před 65. rokem a s pozdním nástupem po 65. roce. První varianta bývá nejčastěji rodově dědičná (familiární) s autozomálně dominantní dědičností, druhá varianta může být jak dědičná, tak sporadická (bez zaznamenaného výskytu v rodině).
- Příznaky: Zpočátku se nemoc projevuje zvýšenou zapomnětlivostí, zmateností, zhoršeným vybavováním slov. Nejvíce je postižena krátkodobá paměť. Nemocný začíná být dezorientovaný, neví, kde je, který je den, dostavují se výkyvy nálady a zvýšená úzkostnost. Choroba postupně doslova vymaže pacientovu identitu a osobnost. V pokročilejších stadiích nemocný často není schopen formulovat věty, může trpět halucinacemi, přestává být soběstačný, neudrží stolici, moč, je zcela odkázán na druhé, potravu mnohdy přijímá pouze sondou zavedenou do žaludku. Postižení Alzheimerovou demencí umírají (v průměru 8–10 let po objevení prvních příznaků) na komplikace plynoucí z nesoběstačnosti a nehybnosti (zápal plic vyvolaný vdechnutím potravy, proleženiny, přidružené infekce apod.).
- Diagnóza Alzheimerovy nemoci je histopatologická. Onemocnění se rozvíjí na základě atrofie mozku (úbytku mozkové tkáně). Pro postiženou mozkovou tkáň je charakteristický vysoký výskyt neurofibrilárních klubek a neuritických plaků tvořených jádrem z β-amyloidových vláken (viz obr. ve Vesmíru 78, 328, 1999/6). Ty se sice vyskytují i ve zdravém stárnoucím mozku, ale jejich množství je výrazně nižší. U Alzheimerovy demence jsou postiženy převážně neurony vytvářející acetylcholin (neuropřenašeč důležitý pro správnou funkci paměti). Patologické změny vyřadí hipokampus a další části spánkového mozkového laloku, které jsou nezbytné pro ukládání paměťové stopy.
- Etiologie: Na vzniku Alzheimerovy nemoci se podílejí jak faktory prostředí (spekuluje se o virech, prionech, toxinech, kovech, špatné výživě, stresu), tak vlivy genetické. Dědičných je asi 40 % případů. Pro vznik geneticky podmíněné formy jsou významné mutace v některém ze čtyř genů. Konkrétně jde o gen pro amyloidový prekurzorový protein na 21. chromozomu, gen pro transmembránový protein presenilin 1 na 14. chromozomu (jeho mutace přispívají k 30–70 % časných onemocnění Alzheimerovou chorobou), gen pro presenilin 2 na 1. chromozomu a gen pro apolipoprotein E na 19. chromozomu (alela tohoto genu r4 se vyskytuje u poloviny případů dědičné Alzheimerovy nemoci s pozdním nástupem). O významu produktů zmíněných genů při rozvoji této nemoci viz též údaje v článku a Vesmír 81, 16, 2002/1.
- Ochranné faktory: Riziko propuknutí choroby podle epidemiologických studií snižují čtyři faktory: užívání protizánětlivých léků (patrně se na vzniku choroby podílejí zánětlivé a imunitní procesy), vyšší vzdělání (jednak zvětšuje mozkovou rezervu, a tím oddaluje nástup příznaků, jednak asi má ochranný vliv), užívání estrogenů u žen a podle rozsáhlé studie z Francie i pití malých dávek červeného vína.
- Léčba Alzheimerovy nemoci je zaměřena na projevy. Jejím cílem je zlepšit oslabenou acetylcholinergní komunikaci mezi neurony – a to buď podáváním „stavebních kamenů“ pro tvorbu acetylcholinu (cholinu, lecitinu), popřípadě látek působících na acetylcholinové receptory (pilocarpinu) nebo zablokováním enzymů rozkládajících acetylcholin v synapsích preparáty, jako jsou fyzostigmin nebo novější tacrin a donepezil. Testují se způsoby jak zabránit vzniku či shlukování β-amyloidu nebo jak již vzniklý amyloid odstranit např. pomocí protilátek.
Ke stažení
 Článek ve formátu PDF [4,06 MB]
Článek ve formátu PDF [4,06 MB]
O autorech
Jan Konvalinka
Jana Peichlová
Doporučujeme

Když bahno teče jako ledovec

Ideologie v mapách, mapy v rukách ideologů 
