










Kvantová chemie a počítače
| 5. 1. 1995Počátkem padedesátých let, vdobě kdy Linus Pauling získal Nobelovu cenu, počítače umožňovaly racionalizaci výpočtů složitých matematických vzorců, které se vyskytují vkvantové teorii. Byla navržena schémata řešení Schrödingerovy rovnice avyvinut matematický aparát vhodný kjejich řešení na počítačích. Roothan formuloval verzi Hartreeovy - Fockovy metody, kde jsou molekulární orbitaly vyjádřeny jako lineární kombinace atomových orbitalů. Řešení soustavy diferenciálních rovnic bylo převedeno na maticový tvar abyl připraven matematický aparát pro rozsáhlé kvantověmechanické výpočty atomové imolekulové struktury. Zároveň byly vypracovány nové algoritmy výpočtu integrálů převoditelných do počítačovéhokódu.

V té době bylo zcela normální, dokonce inezbytné, že uživatelé detailně znali počítač, na kterém pracovali, jehostrojový jazyk, ahlavně všechna jehoomezení. Vývoj programů vyžadoval, aby špičkový kvantový chemik byl ipočítačovým odborníkem, či aby stakovýmto specialistou úzce spolupracoval. Přes
Rozvoj kvantové chemie je úzce svázán srozvojem počítačové technologie. Vprůběhupadesátých let byly vyvinuty přibližné metody, které braly ukaždéhoatomu vúvahupouze jeden atomový orbital, kolmý na rovinu molekuly či jejíhofragmentu. Tyto ο-elektronové metody přispěly kinterpretaci některých fyzikálních vlastností poměrně složitých organických molekul. Všedesátých letech již přibližné metody uvažovaly všechny valenční elektrony, a to jak uorganických, tak u anorganických sloučenin. Od roku 1970 použití výkonných počítačů umožňuje výpočty elektronové struktury větších molekul na ab initio úrovni, tedy metodami, které nepoužívají experimentálně adjustované parametry azahrnují všechny elektrony zkoumanéhosystému. Začínají se provádět teoretické studie dynamiky elementárních chemických reakcí, studie mezimolekulových interakcí, vypracovávají se nové přístupy křešení Schrödingerovy rovnice. Kvantová chemie je výrazně ovlivněna rozvojem experimentálních metod chemické fyziky souvisejícím též sefektivností výpočetní techniky. Tehdejší výpočty byly prováděny na sálových počítačích, scentrální jednotkou umožňující současnou práci více uživatelů napojených pomocí terminálů.
V poslední době se vývoj počítačové techniky rozčlenil do několika větví. Stále existují sálové počítače, nyní samozřejmě výkonnější, apro nejnáročnější typy výpočtů byly vyvinuty takzvané superpočítače. Superpočítač má již několik procesorů, na kterých je možno provádět nezávislé výpočty (tzv. paralelizace),
Dnes je stále více populární používání tzv. pracovních stanic, jejichž výkon se výrazně zvětší sdružováním do skupin. Poměr výkonu a ceny pracovních stanic je velice výhodný a relativně nízká cena umožní i menším pracovním kolektivům vlastnit tyto stanice. Nesmíme zapomenout ani na osobní počítače, z nichž nejnovější typy, vybavené několika procesory jako např. Pentium, již dosahují výkonů, které byly před několika lety doménou superpočítačů. Výkonné osobní počítače i pracovní stanice jsou vybaveny grafickými programy a umožňují znázornění jak dvourozměrných, tak třírozměrných obrazů. Rozmach počítačových sítí dovoluje vzájemně propojit všechny typy počítačů. Lze tedy základní program a data připravit na osobním počítači, náročné výpočty provádět na superpočítači vzdáleném třebas několik set kilometrů a výsledky graficky zpracovávat opět na svém pracovišti.
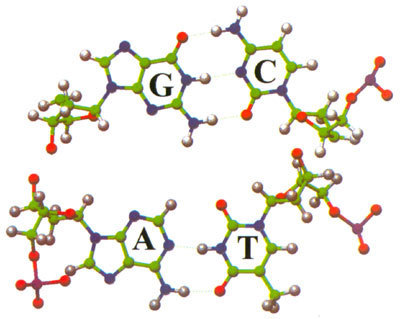
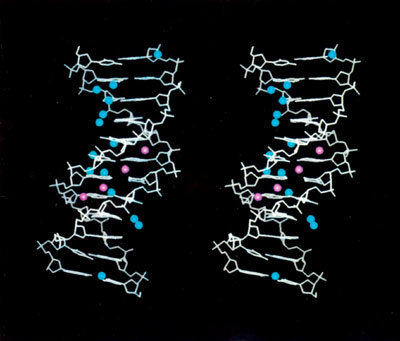
Moderní kvantověmechanické metody slouží k interpretaci mnohých experimentálních výsledků a umožňují modelování dějů v různých oborech chemie i fyziky. Mohou být používány při studiu velkých biologických systémů, jakým je životně důležitá dezoxyribonukleonová kyselina (DNA), i při modelování technologicky významných katalytických procesů. V zájmu osvětlení funkce DNA byly studovány interakce dusíkatých zásad, které jsou její nejdůležitější složkou (vzájemnou polohu těchto bází nám ukazuje obrázek v barevné příloze). Další obrázek, na kterém je znázorněn dekamer DNA, nás přesvědčí o tom, jak velké systémy by měly být v budoucnu studovány. Kvantová mechanika je využívána v programech sloužících k návrhu nových systémů na základě znalostí fragmentů a vazebných vlastností. O náročnosti kvantověchemických programů svědčí i to, že je vyvíjí po řadu let mnoho pracovníků. Např. GAUSSIAN 92, program pro výpočty na ab initio úrovni, má v současnosti 21 spoluautorů a je dále vyvíjen. Finanční náročnost a možnost aplikace samozřejmě vede k tomu, že programy jsou využívány též ke komerčním účelům. Komerční programy obvykle mají snadno ovladatelné uživatelské prostředí, je v nich integrováno několik kvantověmechanických metod a výstupy jsou názorně graficky prezentovány. Přes snadnost obsluhy těchto programů může získané výsledky správně identifikovat pouze kvalifikovaný pracovník.<4.jpgL>
Kvantitativní ab initio metody v kombinaci s různými experimentálními technikami mohou poskytovat nové poznatky o struktuře a molekulárních vlastnostech. S vzrůstající velikostí systémů však výpočetní náročnost strmě roste a tak zde stále nacházejí uplatnění metody, které používají jistá fyzikálně oprávněná zanedbání a experimentální parametry. Všechny tyto kvantověchemické metody umožňují interpretaci základních fyzikálních vlastností, jakými jsou např. přechody v optických spektrech. O tom, ve které části optického spektra látka absorbuje, rozhoduje energie přechodu ze základního stavu do stavu vzbuzeného. Tato energie je dostupná výpočtem a tak můžeme předpovědět, zda látka bude barevná, či nikoliv. Výpočet nábojů na jednotlivých atomech může ukázat, která centra v molekule jsou reaktivní. Výpočty mohou dát velmi dobrou výpověď o geometrické struktuře, vypočtené vazebné vzdálenosti a úhly bývají ve shodě s analýzou na základě X-paprskové difrakce. Pro nové sloučeniny lze na základě výpočtu předpovědět nejpravděpodobnější geometrické uspořádání.
Až doposud jsme hovořili o výpočtech izolovaných systémů. Samostatné molekuly lze nalézt v kosmickém prostoru a je možno je připravit za speciálních podmínek ve vakuu, v přírodě se však vyskytují vždy ve společnosti dalších a jsou jimi do jisté míry ovlivňovány. Molekuly rozpuštěné látky jsou obklopeny molekulami rozpouštědla, chemické reakce se uskutečňují v plynném či kapalném prostředí nebo v pevné fázi. Takovéto systémy, které obsahují velké množství interagujících molekul, nelze popsat přesně. Zde je třeba vytvořit zjednodušený model, který postihuje alespoň základní rysy komplexního systému. Jednou z možných strategií je zjistit co nejpřesnějším způsobem elektronovou strukturu jednotlivých molekul, poté zjistit mezimolekulární interakce a vyjádřit je matematicky pomocí vhodných potenciálů. Celý systém je pak popsán prostřednictvím metody Monte Carlo, molekulárně dynamických metod či statistické termodynamiky. Rozsah článku nám nedovoluje podrobný popis těchto metod, omezme se na konstatování, že využívají poznatků statistiky a kladou velké nároky na výpočetní techniku.
V tomto příspěvku jsme se snažili nastínit to, že úspěšné využívání poznatků kvantové mechaniky úzce souvisí s rozvojem počítačové techniky. Větší účinnost počítačů umožňuje nejen stále přesnější výpočty reálných, i modelových systémů stávajícími metodami, zefektivňování dosavadních programů, ale i formulaci nových metod a přístupů k řešení Schrödingerovy rovnice. Teoretický výzkum však musí být těsně svázán se získáváním nových experimentálních poznatků o fyzikální podstatě přírodních dějů.
Obrázky

O autorovi
Stanislav Záliš
Doporučujeme

Když bahno teče jako ledovec

Ideologie v mapách, mapy v rukách ideologů 
