










Priony
Rozruch a klevety kolem nemoci šílených krav, Creutzfeldtovy-Jakobovy nemoci a nebezpečného hovězího masa již utichly, novináři a televizní komentátoři ztratili o tuto notně vychladlou novinku dávno zájem. Pro podstatně pomalejší rytmus, v němž žije vědecká veřejnost, však události teprve na podzim minulého roku pokročily do další fáze předáním Nobelovy ceny Stanleymu Prusinerovi za objevení a charakterizaci infekčního proteinového agens, všeobecně známého pod názvem prion (proteinaceous infectious particle).
Prusinerovy zásluhy počínají rokem 1972, kdy se rozhodl přijmout a otestovat hypotézu, že těžká neurodegenerativní choroba, na niž zemřel jeden z jeho pacientů, může být způsobena pouze bílkovinami. O deset let později, v roce 1982, Prusiner spolu se svými pracovníky připravil čistý preparát s infekčním agens způsobujícím neurodegenerativní onemocnění křečků a podle všech dokladů preparát obsahoval pouze bílkovinu (viz rámeček 1 ). O dalších patnáct let později již znali v Evropě označení prion snad všichni čtenáři novin a rok po aféře s britským hovězím přibylo Prusinerovo jméno na seznam laureátů Nobelovy ceny.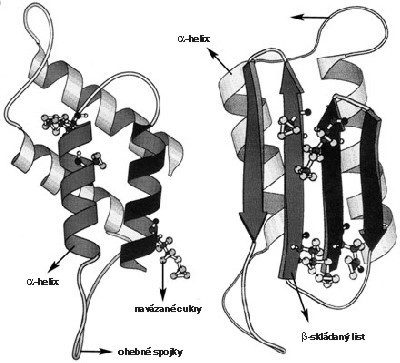
Historie prionů však začala v Anglii 30. let studiem ovčí nemoci scrapie a pokračovala v 50. letech na ostrově Papua Nová Guinea, kde pozoroval D. C. Gajdusek nemoc kuru, přenášenou rituálním pojídáním mozků zemřelých členů kmene. Již tehdy si vědci uvědomovali, že původce nemoci musí patřit do nějaké hodně zvláštní skupiny patogenů. Neobvykle dlouhá inkubační doba, dramatický průběh s podivnými příznaky a netypická epidemiologie, to vše nasvědčovalo možnosti objevení nových infekčních činitelů.
 Dnes jsme samozřejmě v znalostech podstatně pokročili, a to mimo jiné díky systematické práci S. B. Prusinera. Bylo by však předčasné domnívat se, že jediná nezodpovězená otázka, která nám u studia prionů ještě zůstala, se týká léků proti prionovým onemocněním.
Dnes jsme samozřejmě v znalostech podstatně pokročili, a to mimo jiné díky systematické práci S. B. Prusinera. Bylo by však předčasné domnívat se, že jediná nezodpovězená otázka, která nám u studia prionů ještě zůstala, se týká léků proti prionovým onemocněním.
Prionová onemocnění
Jako prionové onemocnění dnes označujeme všechny ty nemoci, které jsou způsobeny abnormálním metabolizmem a konformací, tedy trojrozměrnou stavbou prionového proteinu PrP s jeho následným hromaděním v buňce (viz obrázek). Normální prionový protein zdravé buňky (PrPc – celulární) je u nemocných jedinců změněn na typ se zcela odlišnou trojrozměrnou strukturou (obrázek). Tyto změny způsobují, že PrPsc (Sc znamená scrapie, prionové onemocnění ovcí) je na rozdíl od PrPc nerozpustný a odolný proti enzymům štěpícím proteiny (proteázám). V této téměř nezničitelné podobě se PrPsc hromadí v buňce, ovlivňuje její fungování a v konečném důsledku způsobuje zřejmě degeneraci nervové tkáně.
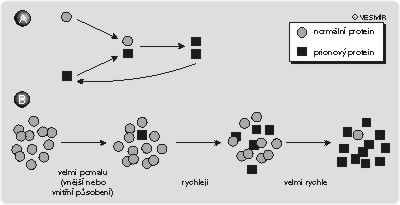
Zmíněné vlastnosti nejsou nijak neobvyklé – proteiny mohou mít různé prostorové uspořádání, které může podstatně ovlivňovat jejich funkci (viz dále). Nejdůležitější a nejpřekvapivější vlastností PrPsc je jeho infekčnost, tedy schopnost jakéhosi „rozmnožování“ a šíření. Tato schopnost je zajištěna velmi neobvyklým způsobem: abnormální protein interaguje s normálním buněčným proteinem PrPc a změní jeho strukturu. Z počáteční dvojice jednoho funkčního a jednoho změněného proteinu vzniknou tedy dva změněné, které se mohou zase oddělit a spojit s dalšími normálními buněčnými proteiny (obrázek). Vinou této „řetězové reakce“ teoreticky stačí vnést do buňky jediný abnormální protein, který bude převádět správné bílkoviny na nesprávné a tak způsobí rozvoj onemocnění. A jen vinou této schopnosti může být nemoc přenosná čistým bílkovinným preparátem (viz rámeček 1 ), ale také hormonálními léčebnými přípravky izolovanými z mozkové tkáně nebo nedostatečně sterilizovanými mozkovými elektrodami.
Infekčnost bílkovinného preparátu je tedy zajištěna vnesením jakéhosi „semene“ (viz obrázek a citaci z Kurta Vonneguta pod článkem), které změní své okolí k obrazu svému. Toto „semeno“ se může objevit ve zdravé buňce třemi různými způsoby. Mezi obyvatelstvem vyvolal paniku první možný způsob: přenos infekčního proteinu prostřednictvím masa, respektive jakéhokoliv biologického materiálu z mrtvol. V mrtvých tělech možná dochází k přeměně PrPc na PrPsc spontánně, a tyto nefunkční proteiny se mohou dostat do mozku živého organizmu po konzumaci kontaminovaného jídla nebo např. léčebným užitím mozkových preparátů. O tom, zda k nákaze dojde, rozhoduje zřejmě podobnost prionového proteinu různých druhů organizmů. Lidský a hovězí PrP jsou si poměrně podobné, takže se navzájem rozeznají a může dojít k řetězové reakci přeměny PrPc na PrPsc.
Prionová neurodegenerativní onemocnění však mohou být i dědičná. K jejich rozvoji dochází u těch jedinců, kteří zdědili poškozený gen PrP, jehož bílkovinný produkt je nestabilní a sám přechází do abnormálního tvaru. Třetí možnost rozvoje prionového onemocnění jsou tzv. náhodná či sporadická onemocnění, kdy v buňkách vznikají priony spontánně nebo vlivem mutace. Tyto případy, jak už jejich název naznačuje, nejsou příliš obvyklé, nicméně jednoznačně dokazují možnost spontánního přechodu PrPc na abnormální formu.
Přenos prionového onemocnění
Priony byly až donedávna jen jakousi zajímavostí, kuriozitou pro zvědavé vědce. Změna, provázená mediálním pobouřením a politickými projevy, nastala po zjištění, že onemocnění např. Creutzfeldtovou-Jakobovou nemocí nemusí být jen tvrdou ranou osudu, ale například trestem za zálibu v krvavých biftecích. Důsledkem tohoto zjištění bylo intenzivní studium přenosu prionového onemocnění mezi různými druhy organizmů. A jak historie už mnohokrát potvrdila, důkladné šťourání do problému přineslo jen řadu dalších otázek.Mezidruhový přenos prionového onemocnění je vždy spojen s prodloužením inkubační doby. Jaká je příčina této tzv. „mezidruhové bariéry přenosu prionového onemocnění“? Pochopitelně hlavní je již zmiňovaná podobnost PrP u zdroje prionů a u jejich příjemce. Mimoto ale bylo zjištěno, že priony tvoří tzv. kmeny. Co se rozumí pod pojmem „prionový kmen“? Jednotlivé prionové bílkoviny, kterými se vědci snaží nakazit myši nebo křečky, se od sebe pravděpodobně odlišují v trojrozměrném uspořádání (dosud neprokázáno), ale rozhodně si uchovávají značný rozdíl v délce inkubační doby, v průběhu onemocnění i ve výběru zasažených míst v mozku. Je možné, že by biologická informace byla dlouhodobě přenášena něčím jiným než nukleovou kyselinou? Jak se uchovává trvalá rozdílnost v inkubačních dobách nemocí jednotlivých prionových kmenů i další „kmenově specifické vlastnosti“? Nehraje zde roli dosud neznámý faktor, například glykosylace (navěšování cukerných zbytků na bílkovinu), nebo dokonce přítomnost nějaké nukleové kyseliny (viz rámeček 1 )?
 To ale není konec všem otázkám, které se při studiu přenosu prionového onemocnění objevily. Některá nová pozorování lze totiž vysvětlit pouze pokud přijmeme existenci ještě dalšího neznámého faktoru X. Tento faktor je pravděpodobně bílkovina a patří zřejmě do skupiny proteinů usnadňujících, či přímo určujících poskládání bílkovin do správného tvaru. Bez jeho pomoci zřejmě nemůže dojít ke změně konformace prionového proteinu a vůbec se nedá vyloučit jeho další role ve vzniku a regulaci hromadění prionů. Jaký má vliv na přenos onemocnění a zda jej vůbec má, to samozřejmě není dosud jasné.
To ale není konec všem otázkám, které se při studiu přenosu prionového onemocnění objevily. Některá nová pozorování lze totiž vysvětlit pouze pokud přijmeme existenci ještě dalšího neznámého faktoru X. Tento faktor je pravděpodobně bílkovina a patří zřejmě do skupiny proteinů usnadňujících, či přímo určujících poskládání bílkovin do správného tvaru. Bez jeho pomoci zřejmě nemůže dojít ke změně konformace prionového proteinu a vůbec se nedá vyloučit jeho další role ve vzniku a regulaci hromadění prionů. Jaký má vliv na přenos onemocnění a zda jej vůbec má, to samozřejmě není dosud jasné.
Zatím jsme mluvili převážně o pozměněné struktuře prionového proteinu, která způsobuje degeneraci nervové tkáně. Funkce buněčného PrPc je ale v celém tomto případě dalším velkým otazníkem (rámeček 2). PrPc je membránový protein přítomný v relativně vysokém množství v nervových buňkách, a bez nějaké zjevné důležité funkce. Myši postrádající tento protein se těší víceméně klidnému životu, stejně jako buňky podobně „opravených“ ovcí.
Priony u kvasinek
Zkusme si pojem prion rozšířit i na jiné proteiny, na všechny ty, které jsou schopny aktivně měnit svou strukturu a v závislosti na ní i svou funkci. Takovéto priony (také označované jako „prionům podobné“, prion-like) byly nedávno k velkému překvapení objeveny u kvasinky Saccharomyces cerevisiae. Získaná data sice nejsou zcela jednoznačná, ani se nedá říci, že by v existenci kvasinkových prionů všichni odborníci uvěřili, nicméně pro pozorované fenomény je to zatím nejjednodušší vysvětlení.Už dost dávno bylo u kvasinek zjištěno, že některé populace buněk mají zvláštní schopnost potlačovat účinek některých typů mutací. Tato vlastnost, označovaná jako PSI+, není dědičná podle mendelovských pravidel, ale získají ji vždy všichni potomci. Z toho jednoznačně vyplývá, že fenotyp PSI+ musí způsobovat nějaký faktor přítomný v cytoplazmě. Dlouho se nedařilo zjistit původce fenomenu PSI. Teprve počátkem let devadesátých se ukázalo, že vlastnost PSI+ je způsobena neobvykle složeným proteinem Sup35p (viz rámeček 3 ).
Jedno z možných vysvětlení pozorovaných jevů předpokládá prionový charakter Sup35p. Podle něj je tento protein schopen zaujmout dvě různá prostorová uspořádání, z nichž každé má poněkud jinou funkci. Prionový protein s porušenou funkcí může interagovat s normálním proteinem a vnutit mu svůj tvar, a tedy i funkci. Důkazů je proto hned několik, i když žádný není přímý. Například buňky PSI+ mohou přejít na fenotyp psi– a zpět zcela spontánně, i když to dělají jen zřídka. Faktory, které způsobují mutace v DNA, nemají na přechod PSI+ – psi– žádný vliv – naopak faktory ničící bílkoviny tento vliv mají. Podobně jako u lidských prionových onemocnění byly i v tomto případě nalezeny takové mutace, které zvyšují pravděpodobnost přechodu ze stavu normálního do stavu změněného. Navíc, abnormální forma proteinu Sup35p vytváří v buňce rozsáhlé agregáty (podobně jako priony v buňkách mozků šílených krav) a nemůže vykonávat svou obvyklou funkci.
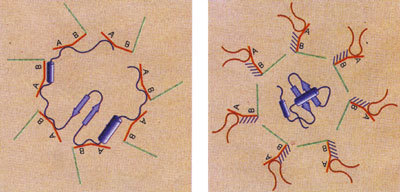
Nicméně dědičnost „prionového“ fenotypu u kvasinek je poněkud ovlivňována i činidly působícími na nukleové kyseliny. Podobné skutečnosti byly zjištěny i u savčích prionů (faktor X, viz výše) a způsobily počátkem 90. let mírnou paniku, že se ta krásná a Nobelovy ceny vskutku hodná teorie bude muset zahodit. Do hry ale místo toho vstoupily další proteiny s neobvyklým jménem chaperony (chaperon – francouzsky gardedáma, obrázek), které zabezpečují správný tvar proteinů vytvářených v buňce.
Chaperony: od tvaru k funkci
Pro správnou funkci bílkovin je důležité nejenom pořadí aminokyselin, které protein tvoří, ale i prostorová struktura, již bílkovina zaujme. Tato struktura je samozřejmě na pořadí aminokyselin závislá, ale nikoliv absolutně. U mnohých bílkovin lze vyměnit řadu aminokyselin, aniž bychom narušili jejich tvar, a tak i jejich funkce zůstává neporušena (pokud samozřejmě nebylo pozměněno nějaké klíčové místo). Jenže průměrná bílkovina se může v prostředí buňky svinout hned několika různými způsoby a jen jeden z nich je správný.Představme si, jak vlastně ke vzniku proteinu v buňce dochází. Ribozomy (supramolekulární útvary tvořené RNA a bílkovinami) překládají podle instrukcí mRNA dědičný text DNA do jazyka proteinů – posouvají se po molekule mRNA a zároveň z nich „vylézá“ nově vznikající bílkovina. Bílkoviny by podle fyzikálních zákonů měly v buňce zaujímat tvar o nejnižší volné energii – tedy energeticky nejstabilnější. Bílkovina ovšem vzniká postupně – a nejstabilnější tvar první poloviny bílkoviny může být zcela odlišný od nejstabilnějšího tvaru bílkoviny celé! Jakmile však první část bílkoviny zaujme nějakou energeticky výhodnou podobu, nemůže se po dosyntentizování jednoduše přesunout do podoby nové – musela by totiž „přelézt energetickou horu“ mimořádně nevýhodného a nestabilního prostorového uspořádání, jakým je rozvinutá bílkovina.
Mnohdy to nevadí – naopak, tvar zaujmutý bílkovinou díky tomu, že byla vytvářena postupně, je ten správný. Jindy však na nově vznikající bílkovinu hned kousek od ribozomu číhají chaperony, které ji složí do toho jediného správného tvaru (obrázek). Chaperony ale dovedou i další věci – například mohou znovu správně poskládat, a tedy i obnovit funkci bílkoviny ničené například teplem nebo jiným denaturačním činidlem. Není tedy divu, že mnoho chaperonů patří do rodiny tzv. heat-shock proteinů (hsp), jejichž tvorba je vyvolána vystavením buněk tepelnému či jinému stresu.
Právě chaperony této veliké proteinové rodiny se podílejí na projevu fenotypu PSI+ u kvasinek. Také lidský prionový protein interaguje s chaperony savčích buněk a v kultuře savčích prionových buněk pravděpodobně dochází ke změnám v jejich tvorbě i funkci. Do kategorie chaperonů zřejmě patří i výše zmiňovaný protein X. Účast těchto proteinů jednoznačně potvrzuje důležitost prostorové struktury PrP v rozvoji prionového onemocnění, i když jejich přesná role zůstává zatím nejasná.
Kolik fenotypů z jednoho genotypu?
Vraťme se zpátky k prionům, k těm unikátním proteinům, které mohou zaujímat dva různé tvary (kdy jeden může získat převahu nad druhým) a tím i dvě funkce. K čemu by se vlastně tato schopnost proteinů mohla v buňce hodit? Je existence prionů dokladem dalšího patogenního činitele, nebo nám spíše ukazuje nový složitý regulační systém eukaryotických buněk?
které mohou zaujímat dva různé tvary (kdy jeden může získat převahu nad druhým) a tím i dvě funkce. K čemu by se vlastně tato schopnost proteinů mohla v buňce hodit? Je existence prionů dokladem dalšího patogenního činitele, nebo nám spíše ukazuje nový složitý regulační systém eukaryotických buněk?
U kvasinek umožňuje PSI+ potlačit negativní účinek náhodných mutací. Tato vlastnost by se ale stala skutečnou výhodou jen tehdy, kdyby se dala podle okolností vypínat či zapínat. A k tomu zřejmě u kvasinky dochází. Abnormální protein se totiž zmnožuje pouze při určitém množství různých chaperonů. Jejich produkce je zase ovlivněna působením tepelného stresu nebo například hladověním. A k hladovění zase může dojít proto, že je nějaký gen nevhodně mutován. Sup35p by v tomto případě mohl být unikátní faktor, který způsobí změny vlastností bez jakékoliv změny genotypů.
 V jednoduché kvasince snad vidíme ve zřetelnějších rysech to, co nám ve složitém soukolí lidského organizmu může unikat. Možná je aktivní a progresivní zaujímání různých tvarů bílkovinami dosud neznámým regulačním mechanizmem jejich funkce, který citlivě reaguje na momentální stav buňky, aniž by předtím vyžadoval změnu záznamu dědičné informace. Vzhledem k téměř byrokratické těžkopádnosti prosazování dědičných změn do DNA by se tato možnost zdála být veskrze praktická. Z tohoto hlediska jeví se prionová onemocnění spíše jako porucha regulací v buňce než nákaza novým nebezpečným patogenem.
V jednoduché kvasince snad vidíme ve zřetelnějších rysech to, co nám ve složitém soukolí lidského organizmu může unikat. Možná je aktivní a progresivní zaujímání různých tvarů bílkovinami dosud neznámým regulačním mechanizmem jejich funkce, který citlivě reaguje na momentální stav buňky, aniž by předtím vyžadoval změnu záznamu dědičné informace. Vzhledem k téměř byrokratické těžkopádnosti prosazování dědičných změn do DNA by se tato možnost zdála být veskrze praktická. Z tohoto hlediska jeví se prionová onemocnění spíše jako porucha regulací v buňce než nákaza novým nebezpečným patogenem.
Vědomostí o prionových proteinech máme už hodně, ale mnohem více nám zůstává nezodpovězených otázek. A ty, jak známo, dosud žádná Nobelova cena nevyřešila.
Obrázky

Citát
Kurt Vonnegut: Kolíbka. Mladá fronta, Praha 1976
[...] Nicméně teoretickým viníkem bylo to, čemu dr. Hoenikker říkal „semeno“. Měl tím na mysli nepatrnou buňku nežádoucí krystalové struktury. Semeno, které se vzalo bůhvíodkud, naučilo atomy zbrusu novému způsobu, jak se seskupovat a vázat, jak krystalizovat, zamrzat.
„A teď si znovu představte dělové koule na trávníku před radnicí nebo pomeranče v bedně,“ vybídl mě [dr. Breed, pozn. red.]. A pomohl mi pochopit, že uspořádání nejspodnější vrstvy dělových koulí či pomerančů určuje, jak bude seskupena a vázána každá vrstva následující. „Spodní vrstva je právě to semeno, které stanoví, jak se zachová každá další dělová koule nebo každý další pomeranč, a tak dál, třeba až do nekonečného množství dělových koulí a pomerančů.“
„A nyní předpokládejme,“ a dr. Breed se zajíkl smíchem, rozradostněn sám sebou, „že by voda mohla krystalizovat, zamrzat několika různými způsoby. Dejme tomu, že druh ledu, na kterém bruslíme a který si dáváme do whisky – můžeme mu říkat led typu 1 – je pouze jedním z několika druhů ledu. Dejme tomu, že voda na Zemi zamrzá vždy pouze jako led typu 1, neboť se nikdy nevyskytlo semeno, které by ji naučilo, jak tvořit led typu 2, led typu 3, led typu 4 ...? A dejme tomu,“ zabušil znovu svou stařeckou rukou do stolu, „že by existoval druh, kterému budeme říkat led typu 9 – krystal tvrdý jako tenhle stůl – s bodem tání řekněme čtyřicet stupňů Celsia, nebo ještě lépe s bodem tání pětapadesát stupňů.“
„Prosím, zatím tomu pořád rozumím,“ řekl jsem.
O přirozenosti prionového infekčního agens
Ne všichni prionologové jsou jednoznačně přesvědčeni, že S. B. Prusinerem obhajovaná proteinová teorie, předpokládající infekčnost čistého proteinu bez přispění nukleových kyselin, může vysvětlit všechny rysy prionových onemocnění. Nikomu se totiž zatím nepodařilo provést definitivní důkaz tohoto tvrzení tedy uměle připravenou abnormální formou prionového proteinu (PrPSc), která nikdy nepřišla do styku s infekčním materiálem, úspěšně nakazit laboratorní zvíře. Zdařilo se zatím jen přeměnit buněčnou zdravou formu prionového proteinu (PrPC) na formu odolnou proti proteázám (enzymům štěpícím bílkoviny), což je charakteristická vlastnost infekční formy prionového proteinu (PrPSc). Jenomže také nikdo dosud neprokázal, že mezi odolností vůči proteázám a infekčností je přímá souvislost, přestože experiment provedený in vitro byl v souladu s některými jevy pozorovanými in vivo (např. s mezidruhovou bariérou či existencí rozdílných prionových kmenů viz text článku).
Infekční materiál izolovaný z mozků zvířat s příznaky prionového onemocnění má několik zvláštních vlastností. Předně jde o nerozpustný agregát, který denaturován a poté znovu renaturován ztrácí schopnost vyvolat u experimentálního zvířete prionové onemocnění. Co se týká nukleových kyselin, popis infekčního prionového agens se omezuje na konstatování, že pokud je v něm obsažena nukleová kyselina, musí být velmi vzácná a kratší než 100 nukleotidů její přítomnost však není vyloučena! Další komplikací proteinové teorie je i fakt, že infekční jednotka (nejnižší počet molekul prionového proteinu nutného k úspěšnému přenosu na organizmus) odpovídá nejméně 100 000. Je snad infekční činitel přenosný pouze v agregované formě, která v sobě skrývá strukturní informaci pro přeměnu zdravé buněčné formy prionového proteinu PrPC na formu infekční PrPSc? Nebo je infekčním činitelem velice vzácná forma prionového proteinu, která se vyskytuje jediná mezi 100 000 obyčejnými? PrPC totiž patří mezi glykosylované membránové proteiny, u nichž jsou na proteinovou kostru připojeny různě pospojované cukerné zbytky, kterých může být v případě PrPC více než 400 různých kombinací. Možná právě varianta v glykosylaci je to, co rozhoduje o infekčnosti. Navíc je prionový protein opatřen poměrně unikátní posttranslační modifikací tzv. GPI kotvou, která může mít nejméně šest různých uspořádání
I přes výrazně převládající víru v proteinovou teorii, v jejímž duchu je prováděna většina experimentů a jejíž hegemonii potvrdilo i letošní udělení Nobelovy ceny, jsou vzácně publikovány a ještě vzácněji citovány i heretické výsledky. V květnu roku 1995 byl například v prestižním Proc. Natl. Acad. Sci. (což je renomovaný časopis americké Národní akademie věd) publikován článek nazvaný provokativně Virové částice jsou nezbytné pro infekční přenos neurodegenerativní Creutzfeldtovy-Jakobovy choroby. V něm se poměrně přesvědčivě dokazuje, že infekčním činitelem jsou částice s podobnými fyzikálně chemickými vlastnostmi, jako mají viry. Autoři prokázali, že prionový infekční materiál obsahuje nukleové kyseliny spolu se specifickými proteiny a že naopak velice čistý prionový protein vykazuje jen minimální infekčnost!
Biologická funkce prionového proteinu - víc otázek než odpovědí
V roce 1984 vědci odhalili hlavní složku prionového infekčního činitele (izolovaného z křeččího mozku a schopného přenést prionové onemocnění na další jedince) a byla to k všeobecnému překvapení bílkovina kódovaná křeččím genomem. Postupně bylo prokázáno, že i v případě dalších prionových onemocnění jiných druhů zvířat a člověka je tato bílkovina, označovaná jako PrP (prionový protein), majoritní složkou infekčního materiálu. Vyskytuje se ve dvou formách: abnormální PrPSc, považovaný za infekční, je nerozpustný, vytváří agregáty a je rezistentní k některým proteázám. Organizmu vlastní, tzv. buněčná forma prionového proteinu PrPC má zcela odlišné vlastnosti: je zakotvena v membráně pomocí tzv. GPI-kotvy, je citlivá k proteolytickému štěpení a vyskytuje se na povrchu řady buněk, v hojném počtu např. v nervové tkáni nebo na některých typech lymfocytů.
Tzv. proteinová teorie předpokládá klíčovou roli prionového proteinu v patogenezi prionových onemocnění. Co je však skutečnou příčinou poruch funkce nervového systému při prionových onemocněních? Je za ně odpovědný PrPSc, který se během onemocnění stále vytváří na úkor PrPC a je schopný při přílišném nahromadění zahubit buňku, popřípadě spustit programovanou buněčnou smrt dalších buněk, aktivovat mozkové astrocyty či některé typy lymfocytů, jak vyplývá z výsledků některých experimentů? Nebo je snad příčinou neurodegenerativních změn snížené množství PrPC v membránách nervových buněk po přeměně v PrPSc?
Přes množství provedených experimentů na tyto otázky dosud nemáme jasnou odpověď. Navíc ani neznáme přesnou fyziologickou funkci PrPC. Lokalizace do vnějšího listu cytoplazmatické membrány by poukazovala na možnou receptorovou, signalizační nebo transportní funkci. In vivo však nebyla nalezena žádná možná látka využívající PrP jako receptor.
V několika laboratořích byly též připraveny myši neschopné syntetizovat PrPC. Výsledky těchto pokusů jsou však rozporné. Nejstarší práce překvapivě nenalezly žádné patologické změny u jedinců bez PrPC, další odhalily poměrně nevýrazné změny v některých mozkových charakteristikách u starých myší v synaptické inhibici či tzv. dlouhodobé potenciaci spojované s krátkodobou pamětí a učením. Nejnovější experimenty provedené s jinými kmeny myší odhalily výrazné změny v chování a neuropatologické změny u jedinců bez PrPC. V jednom z těchto experimentů byla u myší starých 70 týdnů pozorována ztráta Purkyňových buněk, porušení a částečné poruchy koordinace pohybů, v jiném měly myši narušeny cirkadiánní cykly a spánkový režim... Co však bylo všem jedincům bez PrPC společné? Nebylo je možné infikovat prionovým infekčním činitelem, což podporuje představu o nezbytnosti PrPC pro patogenezi prionových onemocnění. Bohužel se však nepotvrdila ideální možnost preventivní genové terapie prionových onemocnění umlčením genu pro PrPC. Takto připravení mutanti totiž v některých experimentech vykazují patologické projevy připomínající samotná prionová onemocnění. Navíc je možné, že skutečné, ještě výraznější změny spojené s nepřítomností prionového proteinu jsou během ontogeneze maskovány proteiny schopnými PrPC nahradit. Jednoznačným testem funkce PrPC a vlivu jeho nedostatku na organizmus by bylo cílené vypnutí syntézy tohoto proteinu v dospělém jedinci, což je díky pokroku molekulární genetiky dnes možné a jistě se na tom usilovně pracuje.
Priony u kvasinek
Sup35p, produkt už dlouho známého genu SUP35, je součástí translačního terminačního faktoru, který ukončuje překládání mRNA na protein. Tento terminační faktor rozeznává na mRNA (která je vždy o něco delší, než by délka proteinu vyžadovala) STOP-kodon, jenž upozorňuje, že předpis pro syntézu nějaké bílkoviny už skončil a dál překládat už nemá smysl. Kvasinky s porušeným terminačním faktorem pročítají i za kodony STOP, takže vznikají bílkoviny prodloužené o nepotřebné aminokyseliny, což není moc praktické. Na druhou stranu, takováto kvasinka má schopnost pročítat i kodony STOP vzniklé náhodnou chybou, takže může potlačit (SUPrimovat) účinky některých typů mutací, což naopak docela praktické je. Kvasinky s fenotypem PSI+ rostou trochu pomaleji než kvasinky normální, a jsou poměrně odolné vůči různým mutagenům. Poněkud méně zřetelné indicie svědčí i pro prionový charakter URE3 determinanty. URE3 fenotyp, který se projevuje schopností kvasinky metabolizovat nejrůznější typy zdrojů dusíku, je zřejmě způsoben neobvyklou konformací regulátoru metabolizmu dusíku kódovaného genem URE2. I v tomto případě kvasinka s fenotypem URE3 roste poněkud pomaleji, ale zase téměř s jakýmkoliv zdrojem dusíku. A i v tomto případě je prionová povaha Ure2p nejjednodušším vysvětlením zjištěných skutečností.
Ke stažení
 Článek ve formátu PDF [641,57 kB]
Článek ve formátu PDF [641,57 kB]
O autorovi
Zuzana Storchová
Doporučujeme

Když bahno teče jako ledovec

Ideologie v mapách, mapy v rukách ideologů 
