










Cystická fibróza
Není snadné určit, zda se Příroda chová k člověku jako laskavý rodič nebo jako nelítostná macecha.
Plinius Starší
Cystická fibróza (CF) je jedním z nejčastějších recesivních autozomálních lidských genetických onemocnění. (U recesivních chorob je „dobrá“ funkční alela [neboli forma] genu dominantní, a proto se u heterozygotů, kteří od jednoho rodiče zdědili „dobrou“ a od druhého rodiče „špatnou“ alelu, nemoc neprojeví, klinické problémy nastávají pouze u homozygotů, kteří získali nefunkční alely od obou rodičů.) Autozomální znamená, že znak není vázán na pohlavní chromozomy.
Rozšíření choroby. Pochází mutace Δ F508 z paleolitu?
Nemoc je rozšířena hlavně v Evropě a v Severní Americe mezi europoidní kavkazskou populací – postiženo je jedno z 2 000 až 4 000 živě narozených dětí. Příčinou choroby jsou změny v molekule CFTR (cystic fibrosis transmembrane conductance regulator). Gen CFTR se u člověka nachází na 7. chromozomu a bylo popsáno již více než 400 různých mutací jeho DNA. Nejčastější je delece kodonu pro aminokyselinu fenylalanin na 508. místě molekuly (Δ F508), která se vyskytuje v 70 – 90 % případů cystické fibrózy v severní Evropě a v Severní Americe a u 50 % nemocných ve středomořské oblasti. Někteří vědci se na základě zeměpisného a etnického rozšíření Δ F508 a podle četnosti změn v některých intronech (intron je část genové DNA, která nekóduje danou bílkovinu) genu CFTR domnívají, že tato delece je velmi stará a že vznikla již před 52 000 lety v populaci geneticky odlišné od dnešních Evropanů, která byla nejspíše příbuzná dnešním Baskům. Další změny genu nejsou tak časté a jsou více zeměpisně a etnicky omezené; české Kelty budou možná zajímat tzv. „keltské“ alely s mutacemi G551D a R117H, vyskytující se přibližně v oblasti, kam dříve zasahovalo keltské osídlení. Přítomnost alely s mutací G551D (záměna glycinu za kyselinu asparagovou na 551. místě) byla tedy prokázána nejen např. v Irsku, Bretani, Walesu anebo ve Skotsku, ale i v České republice (nikoliv však už např. v Rusku).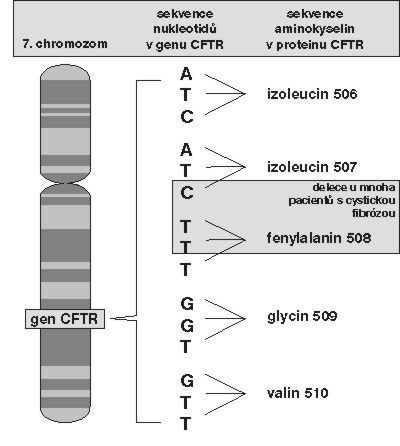

 Vědci se dosud neshodli na tom, proč jsou změny genu pro CFTR v europoidní populaci tak rozšířené. (Uvádí se, že jeden z 23 až 32 lidí je nositelem jedné „špatné“ alely.) Jednou z diskutovaných příčin je zvýhodnění heterozygotů. (Podobná situace je známa např. u srpkovité anemie, kde jsou heterozygoti nesoucí jednu alelu mutovaného hemoglobinu odolnější vůči malárii působené prvokem Plasmodium falciparum.) U cystické fibrózy se uvažuje o tom, že by heterozygoti mohli mít zvýšenou odolnost vůči choleře nebo bronchiálnímu astmatu, ale zde nejsou tyto teorie obecně přijímány.
Vědci se dosud neshodli na tom, proč jsou změny genu pro CFTR v europoidní populaci tak rozšířené. (Uvádí se, že jeden z 23 až 32 lidí je nositelem jedné „špatné“ alely.) Jednou z diskutovaných příčin je zvýhodnění heterozygotů. (Podobná situace je známa např. u srpkovité anemie, kde jsou heterozygoti nesoucí jednu alelu mutovaného hemoglobinu odolnější vůči malárii působené prvokem Plasmodium falciparum.) U cystické fibrózy se uvažuje o tom, že by heterozygoti mohli mít zvýšenou odolnost vůči choleře nebo bronchiálnímu astmatu, ale zde nejsou tyto teorie obecně přijímány.
Proč jsou nemocní s cystickou fibrózou citlivější k infekcím?
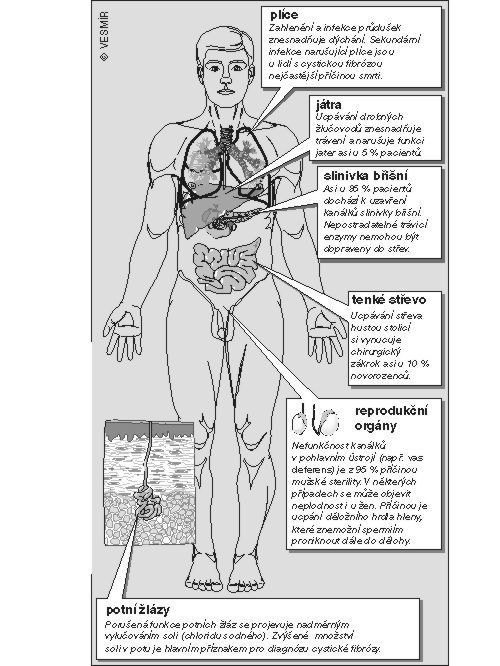
Mutace v genu CFTR vede ke změnám v přenosu chloridových iontů. Gen CFTR kóduje molekulu, která plní funkci chloridové pumpy závislé na cyklickém adenozinmonofosfátu (cAMP). Tato bílkovina se nachází v apikální (z latinského apex – nejvyšší bod) membráně buněk dýchacích cest a dalších buněk epiteliální tkáně. Mutace může protein změnit tak, že vůbec neopustí endoplazmatické retikulum (soustavu kanálků v buňce) a do apikální membrány se nedostane (případ produktu genu s mutací Δ F508), anebo je bílkovina syntetizována normálně, ale vzhledem ke své změněné struktuře nemůže vázat cAMP (G551D). Zvýšení hladiny cAMP v buňce vede ke zvýšené sekreci chloridů do dýchacích cest. Je změněn přenos solí, vody Na+ a dalších iontů, což má za následek nahromadění hlenu v plicích a v zažívacím ústrojí, snížení trávicí i absorpční schopnosti dvanáctníku vinou nedostatečnosti enzymů slinivky břišní, mužskou sterilitu a zvýšení obsahu solí v potu. Nehledě na toto široké spektrum patologických změn je hlavní příčinou smrti poškození plic působené postupně různými chronickými infekcemi a záněty. Nejčastějšími patogeny jsou Staphylococcus aureus, Pseudomonas aeruginosa a Burkholderia cepacia. Není známo, jak zvýšená citlivost k infekcím souvisí s abnormální funkcí CFTR. Předpokládá se, že změněná hladina iontů nějak ovlivňuje vlastnosti hlenové vrstvy a vede k její snížené schopnosti zbavit se vdechovaných mikroorganizmů.Léčba. Antibiotika, rekombinantní DNáza, genová terapie. Myši, které trpí cystickou fibrózou.
Nejběžnějším způsobem léčby je podávání antibiotik s cílem kontroly infekce. V USA a v některých rozvinutých evropských zemích je stále populárnější nový lék – rekombinantní DNáza (komerční název Pulmozyme), která snižuje tloušťku vrstvy s hlenem (sputum) v plicích pacienta. Použití antibiotik zvýšilo délku života pacientů přibližně na 30 let. Rekombinantní DNáza zlepšuje funkce plic, ale vzhledem k poměrně krátké době podávání nelze zatím odhadnout vliv tohoto léku na délku života nemocných. Uvedené terapie však neléčí hlavní příčinu problému, kterou je vadný gen. O to se pokoušejí přístupy genové terapie.Roku 1989 se po dlouhém usilovném výzkumu mnoha vědců, jimž vycházely vstříc i rodiny pacientů, podařilo klonovat gen CFTR. Byl tak učiněn významný krok k lepší diagnostice i k účinnějším léčebným postupům. Následovaly objevy mnoha různých mutací, studium funkce genu (viz výše) i vytvoření myších kmenů, které mají gen pro CFTR různým způsobem změněný. Některé z těchto kmenů trpí podobnými příznaky jako lidé postižení cystickou fibrózou, a tak bylo na těchto modelech možné studovat průběh infekcí a zkoušet genovou terapii. Mutantní myši chované ve standardních podmínkách jsou ve srovnání s jinými kmeny ve zvýšené míře napadány nejméně dvěma z charakteristických CFpatogenů, S. aureus a B. cepacia. Nyní bude velice zajímavé sledovat výsledky studia infekcí nejčastějším z patogenů, P. aeruginosa. Ačkoliv tento mikroorganizmus nenapadá plíce pacientů tak brzy jako S. aureus, je v pozdější fázi onemocnění detektován u 90 % pacientů. Vzhledem k existenci několika subtypů tohoto patogenu je však studium průběhu infekce značně složité.
Pokusy na myších vedly i k nečekanému překvapení. V lidské patologii se již dříve zjistilo, že se klinické příznaky mohou odlišovat i u jedinců se stejnou mutací v genu CFTR. Příčina byla připisována vlivům prostředí. Nyní se ukazuje, že zde hraje úlohu i genetika. Bylo totiž zjištěno, že funkce CFTR je ovlivňována nejméně jedním dalším genem, který se u člověka nachází pravděpodobně na 19. chromozomu.
Po testování na myších proběhlo a v současné době ještě probíhá několik klinických testů genové terapie CF. Byly použity 2 typy léčby: gen CFTR byl vnášen do buněk epitelu plicní tkáně v adenovirových vektorech a v kladně nabitých lipozomech.
 Adenovirus, u primátů častá příčina infekcí horních cest dýchacích, je pro účely genové terapie „zkrocen“. Z jeho DNA jsou odstraněny geny potřebné pro replikaci a je do něho vklonován gen CFTR. Výhody použití těchto vektorů spočívají v tom, že adenovirus přirozeně infikuje buňky epitelu dýchacích cest a že účinnost přenosu genů je relativně vysoká. Tato léčba má však i své stinné stránky. Z neškodného adenovirového vektoru by mohl rekombinací vzniknout replikující se virus. Vlastní bílkoviny adenoviru jsou navíc dost imunogenní a mohou působit zánětlivou reakci (u několika pacientů se zánět skutečně projevil). Prakticky terapie probíhala tak, že gen CTFR v adenovirovém vektoru byl rozprašován do epitelu horních cest dýchacích. Výsledky byly povzbuzující pouze částečně a mnoho kliniků před tímto přístupem varuje. Vytvářejí se proto lepší vektory, kde se odstraňují adenovirové geny, indukující zánětlivou reakci, a zjišťují se optimální léčebná množství (doze) rekombinantního viru.
Adenovirus, u primátů častá příčina infekcí horních cest dýchacích, je pro účely genové terapie „zkrocen“. Z jeho DNA jsou odstraněny geny potřebné pro replikaci a je do něho vklonován gen CFTR. Výhody použití těchto vektorů spočívají v tom, že adenovirus přirozeně infikuje buňky epitelu dýchacích cest a že účinnost přenosu genů je relativně vysoká. Tato léčba má však i své stinné stránky. Z neškodného adenovirového vektoru by mohl rekombinací vzniknout replikující se virus. Vlastní bílkoviny adenoviru jsou navíc dost imunogenní a mohou působit zánětlivou reakci (u několika pacientů se zánět skutečně projevil). Prakticky terapie probíhala tak, že gen CTFR v adenovirovém vektoru byl rozprašován do epitelu horních cest dýchacích. Výsledky byly povzbuzující pouze částečně a mnoho kliniků před tímto přístupem varuje. Vytvářejí se proto lepší vektory, kde se odstraňují adenovirové geny, indukující zánětlivou reakci, a zjišťují se optimální léčebná množství (doze) rekombinantního viru.
Buněčná membrána je záporně nabitá, a proto kladně nabité lipozomy usnadňují průnik DNA do buňky. Výhodou použití lipozomů je méně nežádoucích vedlejších účinků, nevýhodou je menší účinnost přenosu DNA. Pokud byl gen CFTR vnesený do plazmidů podáván v kombinaci s lipozomy, bylo při nejvyšších použitých dávkách rekombinantních plazmidů patrné určité zlepšení klinického stavu pacientů. I zde se dále pracuje na optimalizaci systému.
Uvažuje se i o jiných způsobech přenosu genů, jako je např. cílené směrování genu CFTR, navázaného na vhodný nosič, k receptorům na buňkách epitelu či použití vektorů na bázi parvovirů.
S vývojem účinnější léčby jistě poroste i naděje na úplné vyléčení této hrozné choroby. Anebo se alespoň podaří život pacientů ulehčit.
Literatura
Science 245, 1066, 1989Nature Genetics 12, 280, 1996
Gene Therapy. British Med. Bulletin. Ed. A. M. L. Lever, P. Goodfellow, 51 (1), 1995
Nature Genetics 7, 169, 1995
TIG 12, 81, 1996
Projevy cystické fibrózy a její léčba
Cystická fibróza slinivky břišní (pankreatu) je nejčastějším dědičným onemocněním končívajícím smrtí. Vyskytuje se u jednoho z 2 500 až 3 000 živě narozených dětí. Díky moderní terapii se však v posledních letech pacienti dožívají i dospělosti.Onemocnění je charakterizováno mnohočetným orgánovým postižením. Jednotlivé orgány nejsou postiženy u všech pacientů stejně těžce. 10
O autorovi
Marie Lipoldová
Doporučujeme

Ničí ozon choleru? 

Jak se člověk stává biologem
